As a validation of the cerebral organoid identity, histological sections of a mature (2 month old) cerebral organoid were stained for PAX6 (a marker of dorsal NPCs23) and SATB2 (a marker of mature, postmitotic, upper-layer neurons24). As expected, PAX6+ cells were present at the interior of the organoid, and SATB2+ cells were present in the upper layers (Figure 2). These results support that the cerebral organoids used were indeed dorsal forebrain as specified in the differentiation kit. To establish the dose-dependence of the cerebral organoid transplant system, 2 month old cerebral organoids were injected with increasing numbers of EGFP+ iPSC-derived NPCs. A clear dose-dependence of GFP fluorescence on input cell number was present, with consistent EGFP+ cell patch detection at 10,000 cells and above (Figure 3). The persistence and migration of the transplanted NPCs were next assessed by following the transplanted organoids over time. For this, 50,000 iPSC-derived EGFP+ NPCs were transplanted into 2-3 month old cerebral organoids generated from the same iPSC line. The injected organoids and controls were imaged for EGFP positivity at indicated timepoints over the next 3-4 months. In this transplantation series, we observed persistence of the injected site throughout the 4 month tracking period (Figure 4A). Additional EGFP+ cell patches appeared by 9 days post transplantation and persisted until the study endpoint (3-4 months depending on the organoid), indicating the migration of the cells and integration at their new sites (Figure 4A). At a higher magnification, clear neural morphology was observable with long projections into the organoid (Figure 4B), confirming the integration of the injected cells.
To determine the differentiation status of the injected cells late post injection, the 4 month tracked organoid and its control were fixed, paraffin-embedded, cut to 15 µm thick slices, and mounted onto glass slides. The slices were then processed and stained in either a single round of fluorescent staining (EGFP, TUJ1, NESTIN) or for two consecutive staining cycles to add additional markers (MAP2, GFAP). The initial single-round staining confirmed the presence of EGFP+ cells at the injection site, including a mixture of cells retaining NPC status (NESTIN+TUJ1−) and those that had differentiated towards a neural fate (NESTIN−TUJ1+) (Figure 5). For both the control and injected organoids, very few NESTIN+ NPCs were observed (most, though not all being EGFP+ transplanted NPCs at the site of injection), with a majority of TUJ1+ immature-mature neurons (Figure 5). The two-round staining gave more detail, revealing mature neurons (NESTIN−TUJ1+MAP2+GFAP−) around most of the outer region of the organoid, with areas of immature (NESTIN−TUJ1+MAP2−GFAP−) neurons toward the middle (Figure 6A,B). Astrocytes (NESTIN−TUJ1−MAP2−GFAP+) were present in both the injected and control organoids and were interspersed around the outer edges (Figure 6A,B). The slice for which the two-round staining was performed in the injected organoid showed a small satellite colony of EGFP+ cells far from the injection site that had adopted the phenotype of mature neurons (Figure 6B,C). Some of these appeared to be in close proximity to astrocytes; however, there were no EGFP+ cells with complete overlap to the GFAP staining, suggesting they were adjacent rather than generating the astrocytes themselves (Figure 6B,C).
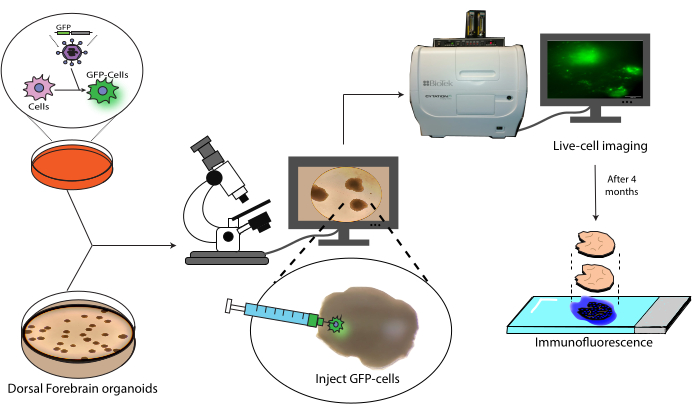
Figure 1: Transplantation model of labeled cells into cerebral organoids. Schematic overview of the generation of labeled cells by lentiviral transduction, their transplantation into cerebral organoids, and tracking by live-cell imaging and immunofluorescence. Abbreviation: GFP = green fluorescent protein. Please click here to view a larger version of this figure.
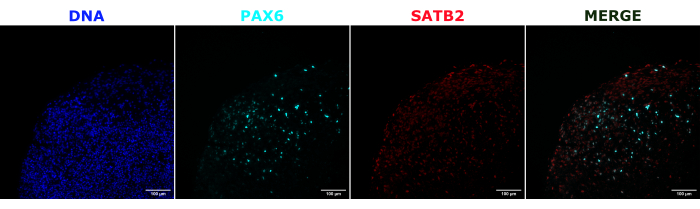
Figure 2: Immunofluorescence of histological sections showing the architecture of early and late organoids. A 2 month old cerebral organoid was fixed, paraffin-embedded, sliced, and stained with PAX6, SATB2, and DAPI. An unstained section was used to set the exposure and integration time to avoid a false-positive signal from autofluorescence. PAX6+ cells were present at the interior of the organoid, while SATB2+ cells were present in the upper layers. Z-stack images were taken every 4.2 µm through the whole 15 µm tissue section. Optical sections were combined using the focus stacking option in the Gen5 software with default options. Scale bars = 100 µm. Abbreviations: PAX6 = paired box 6 protein; SATB2 = special AT-rich sequence-binding protein 2; DAPI = 4',6-diamidino-2-phenylindole. Please click here to view a larger version of this figure.
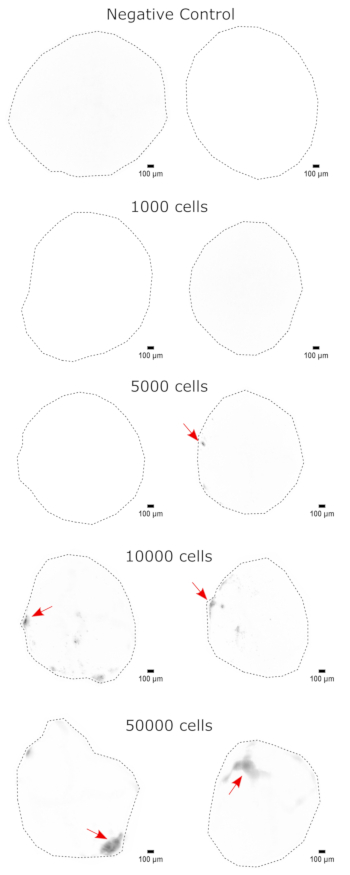
Figure 3: Dose-dependent engraftment of NPCs into cerebral organoids. The organoids were transplanted with 0 (negative control), 1,000, 5,000, 10,000, or 50,000 GFP+ iPSC-derived NPCs. At 1 week post transplantation, the organoids were imaged on a Cytation 5 with a GFP filter cube. The negative control was used to set the exposure and integration time to minimize autofluorescence. Darker colors indicate more EGFP fluorescence relative to the negative control. Scale bars = 100 µm. These are 4x images of the whole organoids. After imaging, rolling-ball background subtraction with a pixel radius of 50 was performed prior to display to correct for the variable background intensity across the organoids. Injection sites identified as the regions of highest engraftment are indicated with a red arrow where engraftment was present. Abbreviations: NPCs = neural progenitor cells; GFP = green fluorescent protein; EGFP = enhanced GFP; iPSC = induced pluripotent stem cell. Please click here to view a larger version of this figure.

Figure 4: Tracking of transplanted cell growth, migration, and persistence with fluorescence live-cell imaging. (A) Control and transplanted (50,000 GFP+ iPSC-derived NPCs) organoids were followed by fluorescence live-cell imaging over the course of 2-4 months from two independent transplant sets. Negative control organoids were used to set the exposure and integration time to minimize the autofluorescence at each time point. EGFP images were captured using a GFP filter cube on the Cytation 5 at indicated times post transplant. Darker colors indicate more EGFP fluorescence relative to the negative control for that time point. Rolling-ball background subtraction with a pixel radius of 50 was performed prior to display to correct for the variable background intensity across the organoids. The organoids were placed in approximately the same orientation at each time point, and the images were rotated for consistency of the display and to clearly show the transplanted cell growth. The injection sites identified as the regions of highest engraftment at the earliest time point are indicated with a red arrow. An example negative control organoid is shown in the bottom of the figure. (B) Example 20x images are shown from engrafted organoids at week 1 and week 15 post transplantation. The local contrast was enhanced prior to display using FIJI to ensure neurite visibility. Scale bars = 100 µm. Abbreviations: NPCs = neural progenitor cells; GFP = green fluorescent protein; EGFP = enhanced GFP; iPSC = induced pluripotent stem cell. Please click here to view a larger version of this figure.
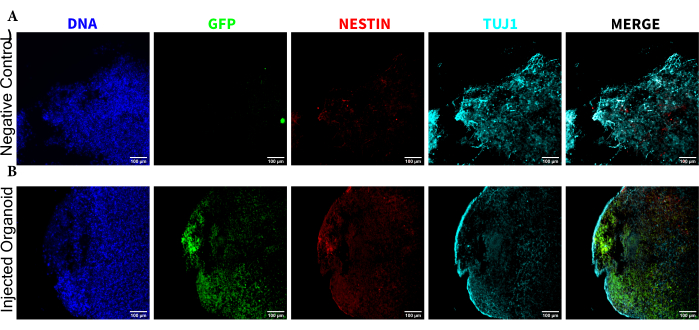
Figure 5: Immunofluorescence of the histological sections revealing the persistence of the transplanted NPCs at the injection site along with migration and neural differentiation. Single-channel fluorescence images from (A) non-injected and (B) transplanted organoids. The overlayed image on the right shows the three channels of interest (NESTIN, TUJ1, and EGFP), but does not include DAPI. The display minimums were set to just exclude the signal from the negative cells (determined for EGFP from the non-injected control and for other channels based on known marker combinations). The display maximums were based on the highest signal observed for that antibody in any cell. The display ranges were kept constant between the non-injected and transplanted organoids to allow for direct comparison. Scale bars = 100 µm. Abbreviations: NPCs = neural progenitor cells; DAPI = 4',6-diamidino-2-phenylindole; TUJ1 = beta-III tubulin; EGFP = enhanced green fluorescent protein. Please click here to view a larger version of this figure.
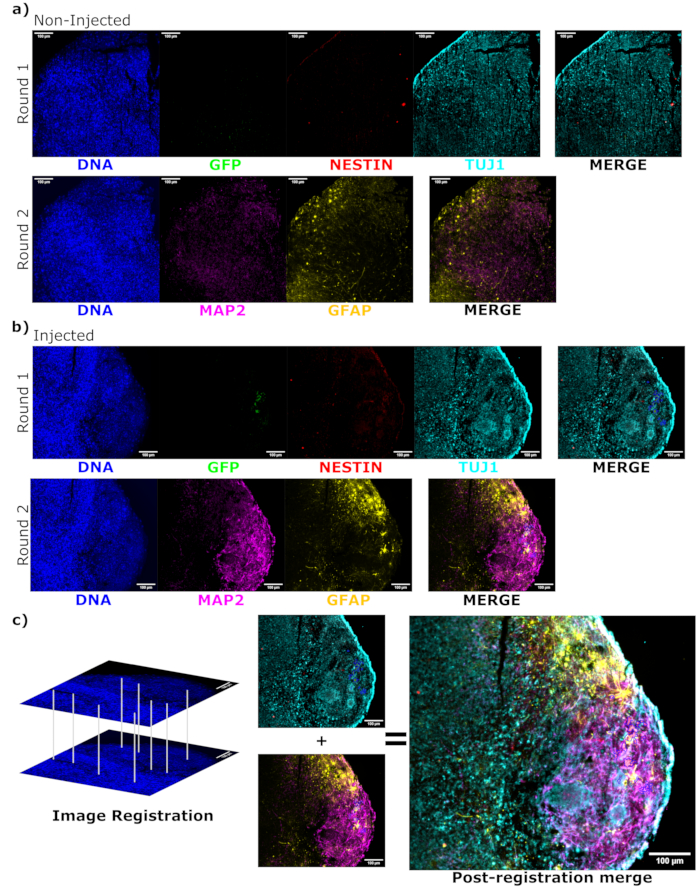
Figure 6: Assessment of engrafted cell differentiation state and localization using cyclic immunofluorescence of the histological sections. Single-channel fluorescence images from the non-injected, (A) age-matched, and (B) transplanted organoids are shown for the first and second round of staining as indicated. The display minimums were set to just exclude the signal from the negative cells (determined for EGFP from the non-injected control and for other channels based on known marker combinations). The display maximums were based on the highest signal observed for that antibody in any cell. The display ranges (and of course, the imaging parameters) were kept constant between the control and injected organoids to allow for direct comparison. Scale bars = 100 µm. An overlayed image (excluding DAPI) is shown for each staining round for each organoid on the right. (B) For the injected organoid where image registration was performed, all images are cropped to the region observed in both the staining rounds. (B,C) For the injected organoid, the EGFP+ cell regions are outlined in blue. A diagram of how DAPI is used to match the features during image registration is shown in (C), followed by an overall merge of the registered image. Abbreviations: DAPI = 4',6-diamidino-2-phenylindole; TUJ1 = beta-III tubulin; EGFP = enhanced green fluorescent protein; MAP2 = microtubule-associated protein 2; GFAP = glial fibrillary acidic protein. Please click here to view a larger version of this figure.
| Clone | Fluorophore | Concentration | |
| Anti-NESTIN | 10C2 | AlexaFluor 594 | 1 in 2,000 |
| Anti-TUBB3 | TUJ1 | AlexaFluor 647 | 1 in 2,000 |
| Anti-GFP | FM264G | AlexaFluor 488 | 1 in 200 |
| Anti-GFAP | SMI 25 | AlexaFluor 594 | 1 in 500 |
| Anti-MAP2 | SMI 52 | AlexaFluor 488 | 1 in 1,000 |
| Anti-PAX6 | O18-1330 | AlexaFluor 647 | 1 in 100 |
| Anti-SATB2 | EPNCIR130A | AlexaFluor 594 | 1 in 500 |
Table 1: Antibody concentrations for staining. Abbreviations: TUBB3 = beta-tubulin III; GFP = green fluorescent protein; GFAP = glial fibrillary acidic protein; MAP2 = microtubule-associated protein 2; PAX6 = paired box 6 protein; SATB2 = special AT-rich sequence-binding protein 2.
