This lysosome proximity labeling proteomics study was conducted in human iPSC-derived neurons to capture the dynamic lysosomal microenvironment in situ in live neurons. Cell morphologies of hiPSCs and hiPSC-derived neurons at different time points are illustrated in Figure 2A. Human iPSCs grow in colonies in E8 medium. Differentiation is initiated by plating iPSCs into doxycycline-containing neuron induction medium. Neurite extensions become more visible each day during the 3 day differentiation. After switching to PLO-coated plates in neuron medium, the neurites form a network between neurons, and axonal extensions become more visible as the neurons reach maturation in 2 weeks. In i3Neurons, the localization of the APEX probe is validated by fluorescence microscopy following rapid APEX activation. Biotinylated proteins are stained using streptavidin (SA) antibody, and lysosomes are stained using anti-LAMP1 antibody. The merged image validates the correct localization of LAMP1-APEX to the bait protein (Figure 2B).
The bead-titration assay is crucial to determine the optimal beads-to-protein ratio so that the amount of streptavidin beads is enough to enrich all the biotinylated proteins but not so excessive to cause serious streptavidin contamination in LC-MS. The optimal volume of beads needed for 50 µg of input protein sample is selected on the basis of where the exponential decay of the curve ends (Figure 3A) As shown in Figure 3A, dot-blot signals from the beads-protein incubation supernatant decreased as more biotinylated proteins were captured with increased amounts of the streptavidin beads. For endogenous LAMP1-APEX samples, 5 µL of streptavidin beads were optimal for 50 µg of input protein (highlighted in Figure 3A). Following enrichment, the amount of protein captured by the streptavidin beads is unknown. Excess proteolytic enzyme (trypsin) can increase enzyme autodigestion, with abundant trypsin peptide peaks in LC-MS. Excessive trypsin can also digest more streptavidin peptides in the samples. Therefore, the amount of protease needed for on-beads digestion should be optimized. Compared to trypsin alone, on-beads digestion with Trypsin/Lys-C mix resulted in the identification of more proteins and peptides and fewer missed cleavages (Figure 3B). Additionally, 1-1.5 µg of protease per 250 µL of streptavidin magnetic beads was optimal to obtain the highest number of identified proteins and the lowest percentage of missed cleavages (Figure 3C). With an optimal beads-to-protein ratio, the same amount of beads should capture the same amount of biotinylated proteins. Therefore, this optimized protease amount can be used for all experiments that enrich biotinylated proteins using the same streptavidin magnetic beads.
Peroxidase-based proximity labeling enzymes are activated by biotin-phenol incubation and brief H2O2 treatment (1 min). This step is a major source of variation in proximity-labeling proteomics. We previously found that normalization to the most abundant, endogenously biotinylated carboxylase, PCCA, can significantly reduce experimental variations, allowing the comparison of proximity labeling proteomics data across different experimental batches (Figure 4)22. For endogenous LAMP1-APEX neurons, the parental line without LAMP1-APEX probe expression was used as a control group. Control neurons were also treated with biotin-phenol and H2O2. The distribution of protein ratios of LAMP1-APEX versus the control group is illustrated in Figure 5A. All the endogenously biotinylated carboxylases were enriched by streptavidin-coated beads but remained unchanged. As shown in the GO-term analysis and protein network analysis (Figure 5B,C), both stable lysosomal membrane proteins and transient lysosomal interactors related to endolysosomal trafficking and transport were enriched in LAMP1-APEX proteomics32,33,34.
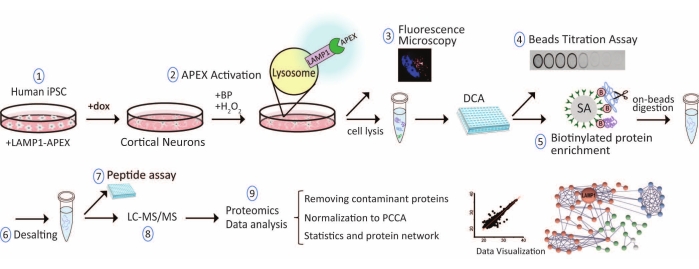
Figure 1: Overall workflow for lysosome proximity labeling proteomics in hiPSC-derived neurons. Abbreviations: hiPSC = human induced pluripotent stem cell; LAMP1 = lysosomal associated membrane protein 1; APEX = ascorbate peroxidase; dox = doxycycline; BP = biotin-phenol; DCA = detergent-compatible protein assay; SA = streptavidin; LC-MS/MS = liquid chromatography-tandem mass spectrometry; PCCA = propionyl-CoA carboxylase, an endogenously biotinylated protein. Please click here to view a larger version of this figure.
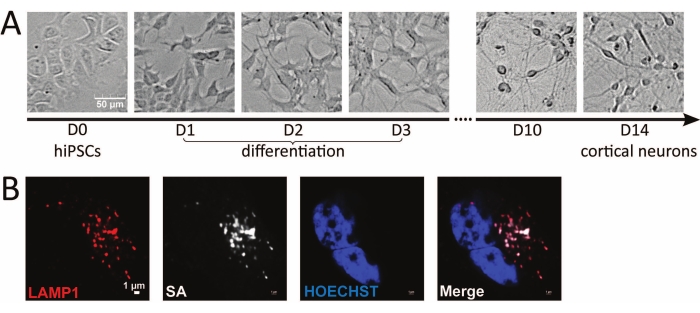
Figure 2: Microscopic imaging of hiPSC-derived neurons and LAMP1-APEX activity. (A) Brightfield microscopy images of different stages of hiPSCs and hiPSC-derived neurons. (B) Fluorescence imaging of LAMP1-APEX activity in the neuron. Biotinylated signals stained against streptavidin colocalize with LAMP1 staining outside the nucleus (HOECHST). Scale bars = (A) 50 µm, (B) 1 µm. Abbreviations: hiPSC = human induced pluripotent stem cell; LAMP1 = lysosomal associated membrane protein 1; APEX = ascorbate peroxidase; SA = streptavidin. Please click here to view a larger version of this figure.
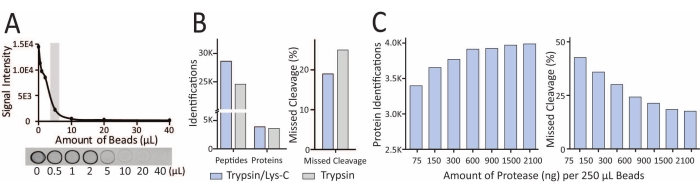
Figure 3: Optimization of beads-to-input protein ratio and enzymatic protein digestion can improve protein identifications and reduce interference. (A) Example of beads-titration assay results from dot-blot assay using 50 µg of input protein and different amounts of streptavidin beads. (B) Trypsin/Lys-C mix resulted in better protein/peptide identification and fewer missed cleavages than trypsin alone. (C) Optimization of the amount of Trypsin/Lys-C for on-beads digestion. This figure has been modified from Frankenfield et al.22. Please click here to view a larger version of this figure.
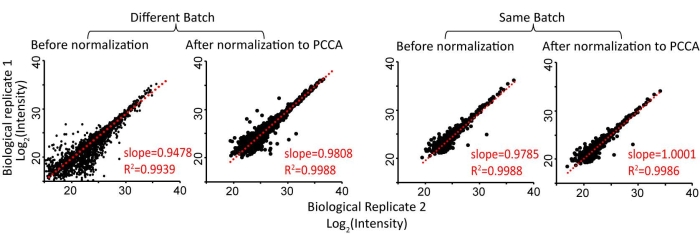
Figure 4: Normalization of proximity labeling proteomics data to an endogenously biotinylated carboxylase, PCCA, can reduce quantification variations among biological replicates. This figure has been modified from Frankenfield et al.22. Abbreviation: PCCA = propionyl-CoA carboxylase. Please click here to view a larger version of this figure.
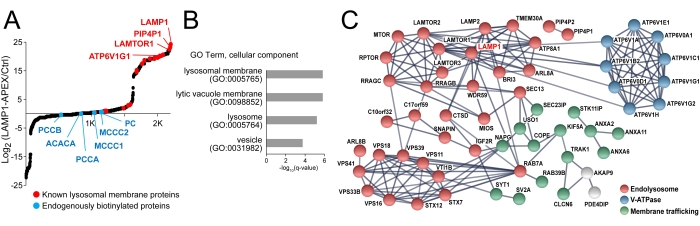
Figure 5: Lysosome proximity labeling proteomics enriched lysosomal membrane proteins and lysosomal interacting proteins in neurons. (A) Scatter plot of protein abundance ratios of LAMP1-APEX versus no APEX control showing enriched lysosomal membrane proteins and unchanged endogenously biotinylated proteins. (B) GO-term analysis of proteomics results proving enriched cellular component at the lysosome. (C) STRING protein network analysis showing that proteins directly interact with the bait protein (LAMP1), lysosomal membrane proteins, and lysosomal interactors such as membrane trafficking proteins. This figure has been modified from Frankenfield et al.22. Abbreviations: LAMP1 = lysosomal associated membrane protein 1; APEX = ascorbate peroxidase; GO = gene ontology. Please click here to view a larger version of this figure.
| Medium/Buffer | Component | Protocol | ||
| Basement membrane matrix (Matrigel) coating solution | 1% basement membrane matrix stock, 99% DMEM/F12 medium | 1.1, 1.2 | ||
| Vitronectin coating solution | 5 μg/mL final concentration in PBS | 1.1 | ||
| E8 complete medium with ROCK inhibitor | 98% E8 medium, 2% E8 supplement, 10 µM Y-27632 or 50 nM Chroman1 | 1.1 | ||
| Neuron Induction medium | 97% DMEM/F12 with HEPES, 1% N2 supplement, 1% non-essential amino acids (NEAA), 1% L-glutamine, 2 μg/mL doxycycline and ROCK inhibitor (10 μM Y-27632 or 5 nM Chroman 1) | 1.2 | ||
| Neuron PLO coating solution | 0.1 mg/mL Poly-L-Ornithine (PLO), 100 mM boric acid, 25 mM sodium tetraborate, 75 mM sodium chloride, 1 M sodium hydroxide | 1.3 | ||
| Neuron medium | 98% cortical neuron medium, 2% B27 supplement, 10 ng/mL brain-derived neurotrophic factor (BDNF), 10 ng/mL glial-derived neurotrophic factor (GDNF), 10 ng/mL NT-3, 0.2 µg/mL Laminin, 2 μg/mL doxycycline | 1.3 | ||
| Quench buffer | 10 mM sodium azide, 10 mM sodium ascorbate, 5 mM TROLOX in PBS | 2.2 | ||
| Cell lysis buffer | 50 mM Tris-HCl, 500 mM NaCl, 0.2% SDS, 1% Triton, 1 mM Tris(2-carboxyethyl)phosphine hydrochloride (TCEP), 10 mM sodium azide, 10 mM sodium ascorbate, 5 mM TROLOX, protease inhibitor cocktail | 2.3 | ||
| TBS-T | 0.05% Tween20, 20 mM Tris, 150 mM NaCl (pH 7.5) | 4.2 | ||
| Buffer A | 2% SDS buffer | 5.2 | ||
| Buffer B | 50 mM Tris-HCl, 500 mM NaCl, 2% Triton-X | 5.6 | ||
| Buffer C | 50 mM Tris-HCl, 250 mM NaCl, 0.5% SDS, 0.5% Triton-X | 5.6 | ||
| Buffer D | 2 M Urea, 50 mM Tris-HCl | 5.6 | ||
Table 1: Compositions of media and buffers used in this protocol.
| Problem | Protocol | Solution/Suggestion | ||||
| iPSC culture peeling off the plate | 1.1 | Increase vitronectin coating concentration or time. | ||||
| Some iPSCs did not differentiate into neurons | 1.2 | Increase cell detachment solution treatment time to completely dissociate iPSCs during d0 differentitation. | ||||
| Neuron culture peeling off the plate | 1.3 | Washing and changing the medium must be gentle and from the side wall of the plate. | ||||
| Proteins do not dissolve completely after acetone precipitation | 2.7 | Reduce drying time of the protein pellet. Increase the volume of lysis buffer and sonicate briefly to help dissolution. | ||||
| Weak streptavidin staining signals | 3 | Increase H2O2 treatment time to be 2-3 s and swirl the plate for even distribution. | ||||
| Low signal of beads titration assay | 4.2 | Wait until membrane is completely dried and add more supernatant to the same spot (can repeat up to 3x) to enhance the signal intensity. | ||||
| Magnetic beads not pelleting towards magnetic rack | 5 | Magnetic bead mobility decreases in non-detergent containing buffer. Higher urea concentration up to 4 M or LC-MS-compatible detergent can be used in wash buffer D. | ||||
| Magnetic beads loss during beads wash | 5 | Increase the waiting time when sample tubes are placed on the magnetic beads (1 min or longer) before taking supernatant from the tubes. | ||||
| Singly charged contamination peaks in LC-MS | 6 | Peptide cleanup not sufficient. Increase washing volumes and times during peptide desalting. | ||||
| Peptide assay low signal | 7 | Resuspend peptide samples in lower volume to increase peptide concentration. | ||||
| Overwhelming streptavidin signals in LC-MS | 8 | Reduce the amount of streptavidin beads. If trypsin peak is also abundant, reduce trypsin amount. | ||||
| Too many nonspecific labeling background | 9 | Streptavidin beads wash was not sufficient. Remove all residual liquid during each washing step. Increase the time and volume for beads wash. | ||||
Table 2: Troubleshooting problems and solutions.
