Multiplexed Analysis of Retinal Gene Expression and Chromatin Accessibility Using scRNA-Seq and scATAC-Seq
Summary
Here, the authors showcase the utility of MULTI-seq for phenotyping and subsequent paired scRNA-seq and scATAC-seq in characterizing the transcriptomic and chromatin accessibility profiles in retina.
Abstract
Powerful next generation sequencing techniques offer robust and comprehensive analysis to investigate how retinal gene regulatory networks function during development and in disease states. Single-cell RNA sequencing allows us to comprehensively profile gene expression changes observed in retinal development and disease at a cellular level, while single-cell ATAC-Seq allows analysis of chromatin accessibility and transcription factor binding to be profiled at similar resolution. Here the use of these techniques in the developing retina is described, and MULTI-Seq is demonstrated, where individual samples are labeled with a modified oligonucleotide-lipid complex, enabling researchers to both increase the scope of individual experiments and substantially reduce costs.
Introduction
Understanding how genes can influence cell fate plays a key role in interrogating processes such as disease and embryonic progression. The complex relationships between transcription factors and their target genes can be grouped in gene regulatory networks. Mounting evidence places these gene regulatory networks at the center of both disease and development across evolutionary lineages1. While previous techniques such as qRT-PCR focused on a single gene or set of genes, the application of high-throughput sequencing technology allows for the profiling of complete cellular transcriptomes.
RNA-seq offers a glimpse into large scale transcriptomics2,3. Single-cell RNA sequencing (scRNA-seq) gives investigators the ability to not only profile transcriptomes but link specific cell types with gene expression profiles4. This is achieved bioinformatically by feeding individual cell profiles into sorting algorithms using known gene markers5. Multiplexing using lipid-tagged indices sequencing (MULTI-seq) offers unprecedented diversity in the number of scRNA-Seq profiles that can be collected6. This lipid based technique differs from other sample indexing techniques such as cell-hashing that rely on the presence of surface antigens and high affinity antibodies instead of plasma membrane integration7. Not only is it now possible to profile gene expression profiles into cell types but different experiments can be combined into a single sequencing library, dramatically lowering the cost of an individual scRNA-seq experiment6. The cost of scRNA-seq may seem prohibitive for use in phenotyping experiments where many different genotypes, conditions or patient samples are analyzed, but multiplexing allows the combination of up to 96 samples in a single library6.
Profiling gene expression via scRNA-seq has not been the only high-throughput sequencing-based technique to revolutionize the current understanding of how molecular mechanisms dictate cell fate. While understanding which gene transcripts are present in a cell enables the identification of cell type, equally important is understanding how genomic organization regulates development and disease progression. Early studies relied on detecting DNase-mediated cleavage of sequences not bound to histones, followed by sequencing of the resulting DNA fragments to identify regions of open chromatin. In contrast, single cell assay for transposon accessible chromatin sequencing (scATAC-seq) allows researchers to probe DNA with a domesticated transposon to readily profile open chromatin at the single nucleotide level8. This has gone through a similar scaling to scRNA-seq and now investigators can identify individual cell types and profile phenotypes across thousands of individual genomes8.
The pairing of scRNA-seq and scATAC-seq has allowed researchers the ability to profile thousands of cells to determine cell populations, genomic organization, and gene regulatory networks in disease models and developmental processes9,10,11,12. Here the authors outline how to first utilize MULTI-seq to condense phenotyping of a myriad of animal models and employ paired scRNA-seq and scATAC-seq to gain a better understanding of the chromatin landscape and regulatory networks in these animal models.
Protocol
The use of animals for these studies was conducted using protocols approved by the Johns Hopkins Animal Care and Use Committee, in compliance with ARRIVE guidelines, and were performed in accordance with relevant guidelines and regulations.
1. MULTI-seq
- Media preparation
- Prepare and equilibrate ovomucoid inhibitor, 10 mg of ovomucoid inhibitor and 10 mg of albumin per mL of Earle's Balanced Salt Solution (EBSS), for 30 min prior to use13. Equilibrate with 95% O2:5% CO2 in an incubator.
- Prepare papain dissociation solution, 20 units/mL papain and 0.005% DNase in EBSS, during this incubation step13.
NOTE: One vial of DNase solution will be sufficient for 5 samples. Additional vials may need to be reconstituted if performing MULTI-seq on more than 5 samples.
- Prepare papain dissociation solution, 20 units/mL papain and 0.005% DNase in EBSS, during this incubation step13.
- Prepare lipid modified oligo barcoding solutions.
- Make a unique barcode dilution for each sample by combining 0.5 µL of 100 µM barcode stock solution with 4.5 µL of Phosphate Buffered Saline (PBS).
- To prepare anchor:barcode solution at 1:1 molar ratio, combine 4.4 µL of 10 µM barcode dilution, 0.9 µL of 50 µM anchor solution, and 16.7 µL of PBS and place on ice. The final concentration for both the barcode strands and anchor strands should be 2 µM.
- To prepare co-anchor solution, combine 0.88 µL of 50 µM co-anchor solution and 21.12 µL of PBS and place on ice. The final concentration of the co-anchor should be 2 µM. Multiply the volumes in this step by 1.1x the number of samples.
- To make 1% BSA in PBS, dissolve 0.1 g of bovine serum albumin (BSA) in 10 mL of PBS and place on ice.
- To make PBS with RNase inhibitor, combine 2.5 µL of 40 U/µL RNase inhibitor with 197.5 µL of PBS and store on ice. Multiply the volumes in this step by 1.1x the number of samples.
- Prepare and equilibrate ovomucoid inhibitor, 10 mg of ovomucoid inhibitor and 10 mg of albumin per mL of Earle's Balanced Salt Solution (EBSS), for 30 min prior to use13. Equilibrate with 95% O2:5% CO2 in an incubator.
- Sample dissociation
- Euthanize animals through cervical dislocation and/or CO2 asphyxia in accordance with institutional IACUC requirements. Method for euthanasia will depend on institutional requirements and model organism used.
- Dissect retina from both eyes in cold PBS under a dissection microscope and transfer to a sterile 1.5 mL microcentrifuge tube using a transfer pipette.
- Transfer 500 µL of the papain solution to a 1.5 mL tube for each sample.
- Place samples in the papain solution and immediately cap the tubes.
- Incubate the tubes containing the samples at 37 °C, 5% CO2 for 30 min. Invert the vials 3 times every 10 minutes. The samples should progressively dissociate with each round of inversions.
- Return the vials to room temperature and triturate each sample 3-4 times with a 1 mL pipette set to 300 µL.
- Allow any pieces of undissociated tissue remaining after trituration to settle.
- Remove the cloudy cell suspension while avoiding any pieces of undissociated tissue and place each sample's cell suspension in a new sterile 15 mL screw-capped tube.
- Centrifuge the cell suspensions at 300 x g for 5 min at room temperature.
- During the centrifugation step, prepare the cell resuspension media.
- Mix 857 µL of EBSS (Vial 1) with 95 µL of reconstituted albumin-ovomucoid inhibitor solution in a sterile 1.5 mL screw-capped tube. Add 48 µL of DNase solution saved from 1.1.1. Multiply the volumes in this step by 1.1x the number of samples.
- Discard the supernatant, careful to retain the cell pellets, and immediately resuspend the cell pellets in 1 mL cell resuspension solution prepared in step 1.2.10.
- Prepare discontinuous density gradient.
- For each sample, add 1.6 mL of ovomucoid inhibitor solution to a new 15 mL sterile screw-capped tube and carefully layer the cell suspension on top. The interface between the two layers of the gradient should be visible but some mixing does not affect results.
- Centrifuge the discontinuous density gradients at 70 x g for 6 min at room temperature. The dissociated cells should pellet at the bottom of the tubes while membrane fragments remain at the interface.
- Discard the supernatant and add 200 µL of PBS to wash the pelleted cells. Centrifuge at 300 x g for 5 min at room temperature.
- Discard the supernatant and resuspend the cells in 180 µL of PBS with RNase inhibitor. Pass the cells through a 40 µm cell strainer into a sterile 1.5 mL microcentrifuge tube.
- Cell barcoding
- Add 20 µL of anchor:barcode solution to each sample and pipette gently to mix. Ensure that each sample receives a unique anchor:barcode solution and incubate on ice for 5 min.
- Prepare the materials required for gel bead-in-emulsion (GEM) generation and barcoding during this incubation14.
- Add 20 µL of co-anchor solution to each sample and pipette gently to mix. Incubate on ice for 5 min.
- Add 1 mL of ice-cold 1% BSA in PBS to each sample and centrifuge the cells at 300 x g for 5 min at 4 °C.
- Remove the supernatant without disturbing the cell pellet and add 400 µL ice cold 1% BSA in PBS. Centrifuge at 300 x g for 5 min at 4 °C.
- Repeat step 1.3.4 for a total of 2 washes. Remove the supernatant and resuspend in 400 µL ice cold 1% BSA in PBS.
- Determine the concentration of cells for each sample using a hemocytometer.
- Determine the total number of cells to load for each sample. Typically, this is the total number of cells desired divided by the number of samples. However, if a biological replicate for one condition was lost, its share can be split amongst the other biological replicates from that condition.
- Divide the number of cells desired for each sample by the cell concentration of that sample calculated in step 3.5 to determine the volume required to load the desired number of cells.
- Combine the volumes calculated in step 3.5.2 from each sample in a new sterile 1.5 mL centrifuge tube. It is prudent to double the volume taken from each sample in case of failure of GEM generation.
- Determine "final" cell concentration of the combined samples using a hemocytometer.
- Add 20 µL of anchor:barcode solution to each sample and pipette gently to mix. Ensure that each sample receives a unique anchor:barcode solution and incubate on ice for 5 min.
- GEM generation and barcoding
- Use combined samples to generate and barcode GEMs14.
- Post GEM-RT Cleanup and cDNA Amplification
- Prepare materials for post GEM reverse transcriptase cleanup and cDNA amplification according to manufacturer's instructions with the following modifications14. Perform post GEM-RT cleanup by size selection14.
- Increase volume of 80% ethanol prepared by 2 mL.
- Prepare cDNA Amplification Mix on ice14. Add 1 µL of 2.5 µM MULTI-seq primer to the cDNA Amplification mix. Vortex for 5 s and centrifuge for 5 s in a benchtop mini centrifuge.
- Perform cDNA amplification14. The cDNA can be stored at 4 °C for up to 72 h at this point.
- Perform cDNA cleanup by size selection14. The endogenous transcript cDNA can be stored at 4 °C for up to 72 h or at -20 °C for up to 4 weeks at this point.
- Do not discard the supernatant from the first round of size selection . This fraction contains the sample barcodes. Transfer the supernatant to a new sterile 1.5 mL microcentrifuge tube.
- Store the supernatant on ice.
- Vortex to resuspend paramagnetic bead-based size selection reagent and add 260 µL paramagnetic bead-based size selection reagent (final 3.2x) and 180 µL of 100% isopropanol (1.8x) to the supernatant. Pipette the mix 10 times and incubate at room temperature for 5 min.
- Place the tube on a magnetic rack and wait for the solution to clear. Then remove and discard the supernatant.
- Add 500 µL of 80% ethanol to the beads and let stand for 30 s. Remove and discard the supernatant.
- Repeat for a total of 2 washes.
- Briefly centrifuge the beads and return to the magnetic rack. Start a timer for 2 min.
- Remove the remaining ethanol with a P10 pipette and air-dry the beads on the magnetic rack for the remainder of the 2 min. Do not exceed 2 min.
- Remove the beads from the magnetic rack and resuspend in 100 µL elution buffer (EB). Pipette thoroughly to resuspend.
- Incubate at room temperature for 2 min.
- Return to the magnetic rack and wait for the solution to clear.
- Transfer the supernatant to a fresh 1.5 mL microcentrifuge tube.
- Repeat steps 1.5.4 to 1.5.12. Halve the volume of buffer EB used in step 1.5.9.
- Determine barcode DNA concentration and size distribution using a dsDNA high sensitivity assay kit and fluorometer and fragment analyzer15,16. The barcode cDNA can be stored at 4 °C for up to 72 h or at -20 °C for up to 4 weeks at this point.
- Prepare materials for post GEM reverse transcriptase cleanup and cDNA amplification according to manufacturer's instructions with the following modifications14. Perform post GEM-RT cleanup by size selection14.
- Barcode library construction
- Prepare 1 mL of 80% ethanol.
- Prepare barcode library PCR master mix in a sterile PCR strip tube: combine 26.25 µL of 2x hot start PCR ready mix, 2.5 µL of 10 µM Universal i5 primer, 2.5 µL of 10 µM RPI primer, 3.5 ng of barcode cDNA (volume determined by concentration from Step 5.14), and enough nuclease-free water to bring the final volume to 50 µL.
- Use a unique RPI primer for each barcode library if multiple MULTI-seq libraries are sequenced together.
- Subject the barcode library master mix to a library preparation PCR: 95 °C for 5 min; 10 cycles of 98 °C for 15 s, 60 °C for 30 s, 72 °C for 30 s; 72 °C for 1 min; and then hold at 4 °C.
- Vortex to resuspend the paramagnetic bead-based size selection reagent.
- Remove the PCR product from the thermocycler and add 80 µL (1.6x) of paramagnetic bead-based size selection reagent. Pipette thoroughly.
- Incubate at room temperature for 5 min. Place on magnet separator in the high position and wait for the solution to clear.
- Remove and discard the supernatant.
- Add 200 µL of 80% ethanol to the beads and allow to stand for 30 s. Remove and discard the supernatant.
- Repeat step 1.6.8 for a total of 2 washes.
- Briefly centrifuge the tube and place on the magnetic separator in the low position. Start a timer for 2 min.
- Remove the remaining ethanol with a P20 pipette and air-dry the beads on the magnetic separator for the remainder of the 2 min. Do not exceed 2 min.
- Remove the tube from the magnetic separator and resuspend the beads in 25 µL of buffer EB. Pipette mix thoroughly to resuspend the beads.
- Incubate for 2 min at room temperature.
- Return to the magnetic separator in the low position and wait for the solution to clear. Transfer the supernatant to a new PCR strip tube.
- Quantify expression library and barcode library concentration and fragment size distribution using adsDNA high sensitivity assay kit and fluorometer and fragment analyzer.
- 3' Gene Expression Library Construction
- Perform 3' gene expression library construction according to manufacturer's instructions14.
- Sequencing
- In addition to the endogenous cDNA library amplified in step 7, submit the barcode library amplified in step 6. For simplicity, submit these libraries separately, or as a cost-effective measure include an aliquot of the sample barcoding material with the endogenous cDNA library. Target between 20,000-50,000 cDNA and 3,000-5,000 barcode reads per cell.
- Convert the resulting sequencing bcl files to fastq files by cellranger mkfastq and count matrices generated from these by cellranger count.
- Deconvolute samples post Cellranger analysis using the deMULTIplex r package available at the MULTI-seq GitHub repository (https://github.com/chris-mcginnis-ucsf/MULTI-seq).
2. Paired scRNA-seq and scATAC-seq
- Flash freeze tissue for scATAC-seq and methanol fixation of retinal cells for scRNA-seq
- Media preparation
- To prepare the solutions for retinal dissociation, repeat steps 1.1.1 and 1.1.4.
- Prepare an ethanol dry ice bath in a small ice bucket.
- Prepare 15 mL of 70% ethanol by adding 10.5 mL of ethanol to 4.5 mL of nuclease-free water.
- Add enough dry ice to the ice bucket (preferably in pellet form) that 15 mL of liquid will rise to just cover the ice. Add the 15 mL of ethanol.
- Place PBS on ice and 1 mL aliquots of 100% methanol in a convenient -20 °C freezer.
- Sample preparation
- Follow steps 1.2.1 and 1.2.2 to isolate retinas; however, this time place each retina into a separate microcentrifuge tube.
- For the samples destined for scATAC-seq, remove any liquid from the microcentrifuge tube using a P200 pipette and immediately cap and place the tube in ethanol-dry ice bath to flash freeze the sample.
- For the samples destined for scRNA-seq, to dissociate the cells, repeat steps 1.2.3 through 1.2.15 on the dissected retina.
- Centrifuge the cells at 300 x g for 3 min at room temperature.
- Remove the supernatant without disrupting the cell pellet.
- Add 1 mL of chilled PBS and gently pipette mix 10 times or until the cells are completely suspended.
- Centrifuge at 300 x g for 5 min at 4 °C.
- Repeat steps 2.1.2.5 to 2.1.2.7 for a total of 2 washes.
- Remove the supernatant without disrupting the cell pellet.
- Add 100 µL of chilled PBS and gently mix 10x or until cells are completely suspended.
- Start vortexing the tube at the lowest speed setting. Add 900 µL chilled methanol (-20 °C) drop by drop while continuing to gently vortex the tube to prevent the cells from clumping.
- Incubate the cells on ice for 15 min.
- Determine the concentration of the fixed cells using a hemocytometer.
- Assess the efficacy of the fixation step using trypan blue. A high fraction of viable cells indicates that more fixation time is required.
- Store the frozen tissue and fixed cells at -80 °C.
- Media preparation
- Nuclei Isolation from flash frozen tissue for scATAC-seq
- Media preparation
- Prepare the lysis dilution buffer by combining 9.77 mL of nuclease-free water, 100 µL of 1 M Tris-HCl (pH 7.4), 100 µL of 1 M NaCl, 30 µL of 1 M MgCl2, and 0.1 g of BSA.
- Prepare the lysis dilution buffer ahead, such as the day before, and store at 4 °C. On the day of the protocol, place on ice.
- Equilibrate 20x nuclei buffer provided by the manufacturer of the scATAC kitfrom -20 °C to room temperature. Vortex and centrifuge briefly.
- Prepare the 1x lysis buffer by incubating 5% Digitonin solution in a 65 °C water bath until the precipitate is dissolved. Then combine 2 mL of lysis dilution buffer, 20 µL of 10% Tween-20, 20 µL of Nonidet P40 Substitute (may alternatively be labeled IGEPAL), and 4 µL of Digitonin.
- Store the 1x lysis buffer on ice.
- To prepare the 0.1x lysis buffer, combine 1.8 mL of lysis dilution buffer with 200 µL of the 1x lysis buffer and store on ice.
- To prepare the wash buffer, combine 2 mL of lysis dilution buffer and 20 µL of 10% Tween-20 and store on ice.
- To prepare the diluted nuclei buffer, combine 950 µL of nuclease-free water with 50 µL of nuclei buffer (20x) and store on ice.
- Nuclei Isolation
- Do not thaw the sample prior to lysis. Add 500 µL of chilled 0.1x lysis buffer to a 1.5 mL microcentrifuge tube containing the frozen sample. Immediately homogenize 15x using a pellet pestle.
- Incubate for 5 min on ice. Prepare the materials required for scATAC transposition during this incubation step17.
- Pipette mix 10x with a 1 mL pipette and incubate for 10 min on ice. Complete preparation of the materials required for scATAC transposition during this incubation step if not already completed. It is unnecessary to prepare more Diluted Nuclei Buffer.
- Add 500 µL of chilled wash buffer to the lysed cells. Pipette mix 5x.
- Pass the suspension through a 70 µm cell strainer into a new 1.5 mL microcentrifuge tube. Filter ~300 µL at a time using a new 70 µm cell strainer each time.
- Pass the collected flowthrough through a 40 µm cell strainer into a new 1.5 mL microcentrifuge tube and store on ice.
- Determine the nuclei concentration using a hemocytometer. Based on the nuclei concentration and the total volume of the cell suspension, calculate the total number of dissociated nuclei.
- Centrifuge at 500 x g for 5 min at 4 °C and remove the supernatant without disrupting the nuclei pellet.
- To create the nuclei stock based on the total nuclei count determined in step 2.2.2.7., resuspend the nuclei in enough diluted nuclei buffer to reach a desired nuclei concentration.
NOTE: Desired nuclei concentration is based on the targeted nuclei recovery for the scATAC-seq experiment and is determined from the Nuclei Concentration Guidelines found in the manufacturer's protocol17. For instance, if 10,000 nuclei are ultimately desired, the desired nuclei concentration for the Nuclei Stock is between 3,080 and 7,700 nuclei/µL. It might be best to aim for the middle of the range in concentration. - Determine the nuclei concentration using a hemocytometer.
- Immediately proceed to scATAC transposition using the prepared nuclei stock17.
- Media preparation
- Rehydration of Methanol Fixed Cells for use in scRNA-Seq
- Media Preparation
- To prepare the wash/dilution buffer, dissolve 0.01 g of BSA in 1 mL of PBS. Then add 12.5 µL of 40 U/µL RNase Inhibitor and mix gently. Store on ice.
- Cell Rehydration
- Centrifuge methanol fixed cells in a 1.5 mL microcentrifuge tube at 3000 x g for 10 min at 4 °C. Then remove the supernatant.
- Add 200 µL chilled wash/dilution buffer to the microcentrifuge tube. Centrifuge at 300 x g for 10 min at 4 °C and remove the supernatant. Repeat for a total of 2 washes.
NOTE: Prepare the materials required for GEM generation and barcoding during the previous two steps14. - To prepare the cell stock, based on the total cell number calculated in 2.1.2.12., resuspend the cells in enough wash/dilution buffer to achieve the desired cell concentration. Preferred cell concentrations are between 700 and 1200 cells/µL.
- Determine the cell concentration of the cell stock using a hemocytometer.
- Immediately proceed to GEM generation and barcoding using the cell stock14.
- Sequencing
- When performing time course analysis (as in development or disease), sequence multiple samples in a multiplexed run (i.e., multiple libraries on a single flow cell) to cut down on technical variation.
- Media Preparation
Representative Results
This workflow lays out a strategy for investigation of developmental phenotypes and regulatory processes using single cell sequencing. MULTI-seq sample multiplexing enables an initial low-cost phenotyping assay while paired collection and fixation of samples for scRNA-seq and scATAC-seq allows for more in-depth investigation (Figure 1).
MULTI-seq barcoding enables the combined sequencing of multiple samples and their subsequent computational deconvolution. The sample of origin can be determined for each cell based on their barcode expression (Figure 2A). These combined samples can be analyzed as a single dataset for the purposes of cell clustering and cell type identification (Figure 2B). Because each cell is barcoded before GEM generation, cell doublets will have a high probability of showing expression for multiple MULTI-seq barcodes and a majority of doublets can therefore be identified and removed prior to clustering and cell type identification (Figure 2C). Increasing the number of cells used in the GEM generation step will increase the proportion of doublets found. scATAC-seq can be used to generate a dataset with cell types to match those found by scRNA-seq (Figure 2D). The pairing of scRNA-seq gene expression and scATAC-seq DNA accessibility information enables the reconstruction of gene regulatory networks.
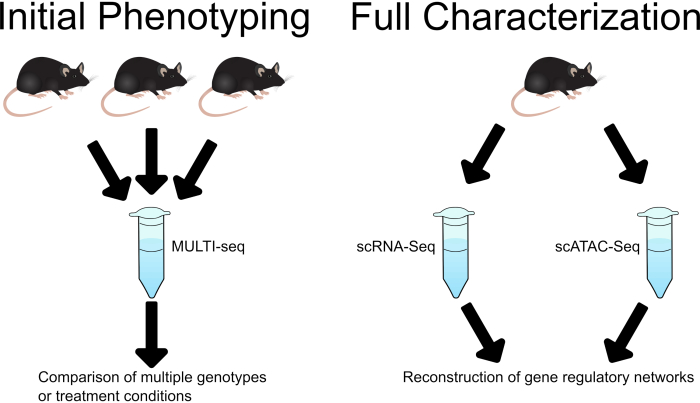
Figure 1: Schematic demonstrating the use of MULTI-Seq in initial analysis, followed by separate scRNA-Seq and scATAC-Seq analysis in in-depth characterization of phenotypes, treatments, or disease states of interest. Please click here to view a larger version of this figure.
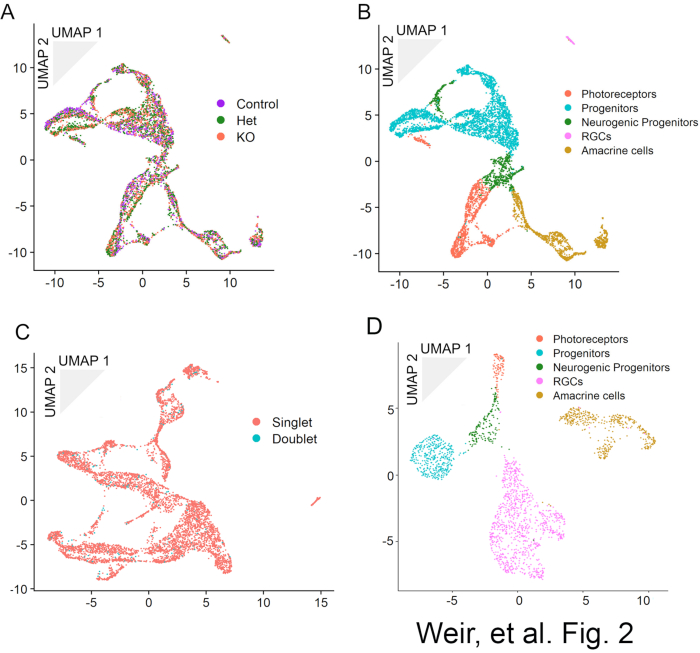
Figure 2: UMAP dimensional reduction representations of MULTI-seq data for an allelic series of P0 Sstr2 knockout mice demonstrating (a) the deconvolution of genotype for each cell in the dataset and (b) the identification of cell types in the dataset. Overloading cells during the GEM generation and barcoding step will result in an increase in cell doublets like those as seen in (c), which shows the data from (a) and (b) before doublet removal and reclustering. In (d), scATAC-Seq data from GFP-positive cells obtained at E16 from retinal explants electroporated with a GFP-expressing control plasmid at E14. Cell types are annotated based on accessibility of cell type-specific genes. This figure has been modified from Weir, K., Kim, D. W., Blackshaw, S. Regulation of retinal neurogenesis by somatostatin signaling. bioRxiv 2020.09.26.314104 (2020) doi:10.1101/2020.09.26.31410418 and original, unpublished data. Please click here to view a larger version of this figure.
Discussion
The power of MULTI-seq stems from seamless integration of data from multiple experimental conditions or models and the enormous benefit in terms of cost and limiting batch effects. Utilizing MULTI-seq offers a laboratory unprecedented phenotyping depth. Non-genetic multiplexing methods such as cell hashing or nuclei hashing opened the door to multiplexed samples through the use of barcoded antibodies7,19,20. However, this relies on the availability of high affinity antibodies that recognize surface proteins expressed on cells or their nuclei, which will not be possible if these antibodies are unavailable or the cells do not express appropriate cell surface or nuclear antigens7. Because MULTI-seq utilizes lipid-modified oligo barcodes to stably incorporate into the cells or their nuclei, it allows researchers to gather transcriptomic data from up to 96 fresh or fixed samples in a cheaper and more broadly applicable manner6.
Following up on the initial MULTI-Seq phenotyping with paired scRNA-Seq and scATAC-Seq is suggested and showcased to gain an understanding of the genomic organization that coincides with the transcriptomic data1. This not only gives an idea of the heterochromatin and euchromatin regions but also valuable understanding of the transcription factor networks driving gene expression. A multi-omic approach can be used to reveal the dynamic chromatin changes that take place at key points of cell fate decisions and determine cellular trajectories in development and disease1,21,22. This can be accomplished through bioinformatically subjecting the data to pseudotime, cis-regulatory interactions and footprinting analysis5,23,24,25. Fixing the samples and sequencing multiple in a multiplexed run reduces sources of batch effect, enabling comparison across samples such as through a time course experiment.The number of tissue samples required depends on the scope of the experiment. When examining the phenotypes associated with embryonic time points, single retinas are often sufficient and can provide hundreds of thousands of cells. A single retina may not provide enough cells in more complex experimental schemes: ex vivo electroporations of retinal explants, probing for rare cell populations, or genetic models with insufficient CRE activation. While a single retina may provide a few hundred cells, these will not capture the full tissue complexity. For such experiments, optimization will be required based on a researcher's needs. Utilizing these techniques, one can gain insight into how the genes analyzed in MULTI-seq are regulated during dynamic processes such as development and disease.
Regulation of gene regulatory networks lies at the heart of understanding cellular processes and how they contribute to development and disease1,26,27. The workflow presented here can be used to identify these gene regulatory networks in specific cell types. However, this protocol has been optimized for use with mouse retinal tissue. Optimization of various steps, such as lysis or dissociation time, centrifugation speeds/times, and number of filtration steps may be required to maximize the number of cells or nuclei and minimize the cell debris in cell suspensions from other tissue types, ex vivo samples, or species, whether samples are fresh, frozen, or fixed in methanol. Methanol fixation time may need to be increased if samples show high levels of viable cells with trypan blue staining. The MULTI-seq technique introduces many additional wash steps over traditional scRNA-seq. To avoid the accidental disposal of valuable cells or DNA, it is prudent to optimize centrifugation speeds and maintain supernatants that would normally be discarded in the cell barcoding, post-GEM RT cleanup, and barcode library construction steps on ice until that step has been verified to be successful. One limitation to MULTI-seq is the limited number of cells, and therefore samples, that can be sequenced from a single well in the GEM generation step. It is recommended to not try to excessively overload cells during this step to avoid a substantial increase in doublet cell capture. Avoid loading a GEM well with more than 20,000 cells. Rather, multiple wells can be prepared from a single combined suspension and multiplexed during sequencing. This will require preparing a large enough volume of cell suspension for GEM generation and the addition of replicates to steps 5, 6, and 7 and will increase the cost of sequencing as more total reads are needed for more cells. With proper optimization, this workflow will enable cost and time efficient identification of cell type-specific phenotypes and gene regulatory networks.
Divulgations
The authors have nothing to disclose.
Acknowledgements
We thank Linda Orzolek from the Johns Hopkins Transcriptomics and Deep Sequencing Core for help in sequencing the produced libraries and Lizhi Jiang for performing the ex vivo retinal explants.
Materials
| 10 µL, 200 µL, 1000 µL pipette filter tips | |||
| 10% Tween 20 | Bio-Rad | 1662404 | |
| 100 µM Barcode Solution | Request from Gartner lab | https://docs.google.com/forms/d/1bAzXFEvDEJse_cMvSUe_yDaP rJpAau4IPx8m5pauj3w/viewform?ts=5c47a897&edit_requested =true |
|
| 100% Ethanol | Millipore Sigma | E7023-500ML | |
| 100% Methanol | Millipore Sigma | 322415-100ML | |
| 10x Chip Holder | 10x Genomics | 1000195 | |
| 10x Chromium controller & Accessory Kit | 10x Genomics | PN-120263 | |
| 15mL Centrifuge Tube | Quality Biological | P886-229411 | |
| 40 µm FlowMi Cell Strainer | Bel-Art | H13680-0040 | |
| 50 µM Anchor Solution | Sigma or request from Gartner lab | https://docs.google.com/forms/d/1bAzXFEvDEJse_cMvSUe_yDaP rJpAau4IPx8m5pauj3w/viewform?ts=5c47a897&edit_requested =true |
|
| 50 µM Co-Anchor Solution | Sigma or request from Gartner lab | https://docs.google.com/forms/d/1bAzXFEvDEJse_cMvSUe_yDaP rJpAau4IPx8m5pauj3w/viewform?ts=5c47a897&edit_requested =true |
|
| 5200 Fragment Analyzer system | Agilent | M5310AA | |
| 70 um FlowMi cell strainer | Bel-Art | H13680-0070 | |
| Allegra X-12R Centrifuge | VWR | BK392302 | |
| Bovine Serum Albumin | Sigma-Aldrich | A9647 | |
| Chromium Next GEM Chip G | 10x Genomics | PN-1000120 | |
| Chromium Next GEM Chip H | 10x Genomics | PN-1000161 | |
| Chromium Next Gem Single Cell ATAC Reagent Kit v1.1 | 10x Genomics | PN-1000175 | |
| Chromium Single Cell 3' GEM, Library & Gel Bead Kit v3.1 | 10x Genomics | PN-1000121 | |
| Digitonin | Fisher Scientific | BN2006 | |
| Dissection microscope | Leica | ||
| DNA LoBind Tubes, 1.5 mL | Eppendorf | 22431021 | |
| Dry Ice | |||
| EVA Foam Ice Pan | Tequipment | 04393-54 | |
| FA 12-Capillary Array Short, 33 cm | Agilent | A2300-1250-3355 | |
| Fisherbrand Isotemp Water Bath | Fisher Scientific | 15-460-20Q | |
| Forma CO2 Water Jacketed Incubator | ThermoFisher Scientific | 3110 | |
| Glycerol 50% Aqueous solution | Ricca Chemical Company | 3290-32 | |
| Hausser Scientific Bright-Line Counting Chamber | Fisher Scientific | 02-671-51B | |
| Illumina NextSeq or NovaSeq | Illumina | ||
| Kapa Hifi Hotstart ReadyMix | HiFi | 7958927001 | |
| Low TE Buffer | Quality Biological | 351-324-721 | |
| Magnesium Chloride Solution 1 M | Sigma-Aldrich | M1028 | |
| Magnetic Separator Rack for 1.5 mL tubes | Millipore Sigma | 20-400 | |
| Magnetic Separator Rack for 200 µL tubes | 10x Genomics | NC1469069 | |
| MULTI-seq Primer | Sigma or IDT | See sequence list | |
| MyFuge Mini Centrifuge | Benchmark Scientific | C1008 | |
| Nonidet P40 Substitute | Sigma-Aldrich | 74385 | |
| Nuclease-free water | Fisher Scientific | AM9937 | |
| P2, P10, P20, P200, P1000 micropipettes | Eppendorf | ||
| Papain Dissociation System | Worthington Biochemical Corporation | LK003150 | |
| PBS pH 7.4 (1X) | Fisher Scientific | 10010-023 | |
| Qiagen Buffer EB | Qiagen | 19086 | |
| Refridgerated Centrifuge 5424 R | Eppendorf | 2231000655 | |
| RNase-free Disposable Pellet Pestles | Fisher Scientific | 12-141-368 | |
| RNasin Plus RNase Inhibitor | Promega | N2615 | |
| RPI primer | Sigma or IDT | See sequence list | |
| Single Index Kit N, Set A | 10x Genomics | PN-1000212 | |
| Single Index Kit T Set A | 10x Genomics | PN-1000213 | |
| Sodium Chloride Solution 5 M | Sigma-Aldrich | 59222C | |
| SPRIselect Reagent Kit | Beckman Coulter | B23318 | |
| Standard Disposable Transfer Pipettes | Fisher Scientific | 13-711-7M | |
| TempAssure PCR 8-tube strip | USA Scientific | 1402-4700 | |
| Trizma Hydrochloride Solution, pH 7.4 | Sigma-Aldrich | T2194 | |
| Trypan Blue Solution, 0.4% (w/v) | Corning | 25-900-CI | |
| Universal I5 primer | Sigma or IDT | See sequence list | |
| Veriti Thermal Cycler | Applied Biosystems | 4375786 | |
| Vortex Mixer | VWR | 10153-838 |
References
- Hoang, T., et al. Gene regulatory networks controlling vertebrate retinal regeneration. Science. 370, (2020).
- Nagalakshmi, U., et al. The Transcriptional Landscape of the Yeast Genome Defined by RNA Sequencing. Science. 320, 1344-1349 (2008).
- Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L., Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature Methods. 5, 621-628 (2008).
- Hwang, B., Lee, J. H., Bang, D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Experimental & Molecular Medicine. 50, 96 (2018).
- Butler, A., Hoffman, P., Smibert, P., Papalexi, E., Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nature Biotechnology. 36, 411-420 (2018).
- McGinnis, C. S., et al. MULTI-seq: sample multiplexing for single-cell RNA sequencing using lipid-tagged indices. Nature Methods. 16, 619-626 (2019).
- Stoeckius, M., et al. Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biology. 19, 224 (2018).
- Chen, X., Miragaia, R. J., Natarajan, K. N., Teichmann, S. A. A rapid and robust method for single cell chromatin accessibility profiling. Nature Communications. 9, 5345 (2018).
- Hoang, T., et al. Gene regulatory networks controlling vertebrate retinal regeneration. Science. , 8598 (2020).
- Clark, B. S., et al. Single-Cell RNA-Seq Analysis of Retinal Development Identifies NFI Factors as Regulating Mitotic Exit and Late-Born Cell Specification. Neuron. 102, 1111-1126 (2019).
- Zheng, Y., et al. A human circulating immune cell landscape in aging and COVID-19. Protein Cell. 11, 740-770 (2020).
- Satpathy, A. T., et al. Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nature Biotechnology. 37, 925-936 (2019).
- Worthington Biochemical Corporation. . Worthington Biochemical Corporation. Papain Dissociation System. , (2020).
- 10x Genomics. . Chromium Single Cell 3′ Reagent Kits v3 User Guide. , (2020).
- Agilent. . DNF-468 HS Genomic DNA 50 kb Kit Quick Guide for Fragment Analyzer Systems. , (2015).
- ThermoFisher Scientific. Qubit dsDNA HS Assay Kits. ThermoFisher Scientific. , (2015).
- 10x Genomics. . Chromium Single Cell ATAC Reagent Kits User Guide (v1.1 Chemistry). , (2020).
- Weir, K., Kim, D. W., Blackshaw, S. Regulation of retinal neurogenesis by somatostatin signaling. bioRxiv. , (2020).
- Stoeckius, M., et al. Simultaneous epitope and transcriptome measurement in single cells. Nature Methods. 14, 865-868 (2017).
- Gaublomme, J. T., et al. Nuclei multiplexing with barcoded antibodies for single-nucleus genomics. Nature Communications. 10, 2907 (2019).
- Ma, S., et al. Chromatin Potential Identified by Shared Single-Cell Profiling of RNA and Chromatin. Cell. , (2020).
- Buenrostro, J. D., et al. Integrated Single-Cell Analysis Maps the Continuous Regulatory Landscape of Human Hematopoietic Differentiation. Cell. 173, 1535-1548 (2018).
- Pliner, H. A., et al. Cicero Predicts cis-Regulatory DNA Interactions from Single-Cell Chromatin Accessibility Data. Molecular Cell. 71, 858-871 (2018).
- Granja, J. M., et al. ArchR: An integrative and scalable software package for single-cell chromatin accessibility analysis. BioRxiv. , (2020).
- Stuart, T., et al. Comprehensive Integration of Single-Cell Data. Cell. 177, 1888-1902 (2019).
- METABRIC Group. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 486, 346-352 (2012).
- Izadi, F. Differential Connectivity in Colorectal Cancer Gene Expression Network. Iranian Biomedical Journal. 23, 34-46 (2019).
