This protocol provides detailed steps for obtaining gene knock-out lines of H. armigera using CRISPR/Cas9 technology. The representative results obtained by this protocol are summarized for gDNA selection, embryo collection and injection, insect rearing, and mutant detection.
In this study, the target site of our gene of interest was located in its second exon (Figure 2A). This site was highly conserved, and the target band fragment of synthesized sgRNA was confirmed using agarose gel electrophoresis (Figure 2B,C,D).
The male and female moths were initially reared in separate net cages to prevent mating ahead of schedule and to ensure a sufficient quantity of embryos as much as possible. In general, a total number of 300 fertilized eggs were collected and were immediately injected with the sgRNA/Cas9 protein mixture (300-500 ng/µL of sgRNA, 200 ng/µL of Cas9 protein) at the one-cell stage. The injection volume was about one-tenth that of the embryos. After microinjection, the embryos were reared as described in section 4, and 40%-60% of injected embryos survived.
The mutant detection of a single sgRNA target was performed by sequencing the PCR products from G1 parental adults (Figure 6B). We also tested the effectiveness of using non-overlapping sgRNA pairs across different exons. The large deletion of the mutants (Figure 6C,D) can be easily distinguished from wild type bands (Figure 6A).
The mutation rate calculated in this protocol was 87.50% when 16 individuals are randomly tested, indicating that this protocol was highly-efficient. Gene knockout results were shown in several genotypes, but the majority of mutants identified from our screening were -2 bp type. Mutations resulted in the premature termination of protein translation in the genome, which subsequently led to the loss of gene function.
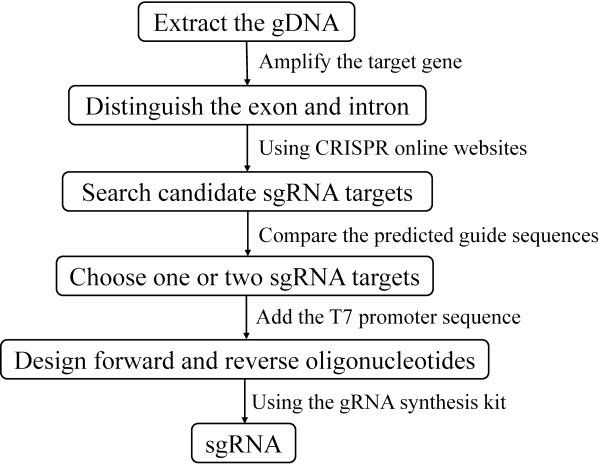
Figure 1: The flowchart for the preparation of sgRNA. Please click here to view a larger version of this figure.
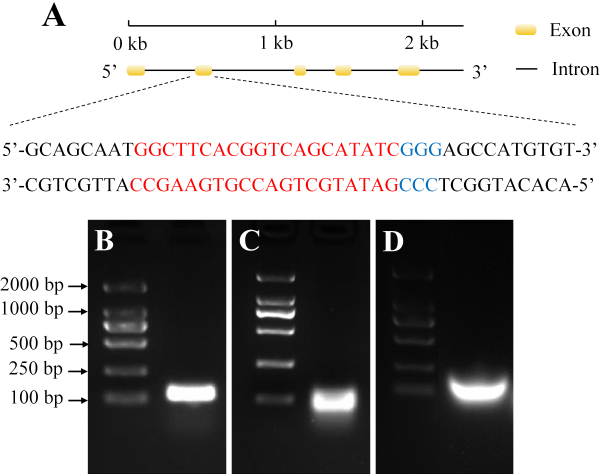
Figure 2: Selection and synthesis of target sgRNAs from H. armigera. (A) The yellow domain represents the exon, while the black line represents the intron. The red sequences indicate the target sequence, and the blue sequences indicate the protospacer adjacent motif (PAM). (B) PCR assembly of the sgRNA DNA template. (C) The in vitro transcription product. (D) Purification of sgRNA. Please click here to view a larger version of this figure.

Figure 3: Embryo collection. (A) A net cage covered with black cloth. The male and female moths of H. armigera were mating. (B) The microscope slide without embryos. (C) The microscope slide containing 50-100 embryos on pieces of black cloth. Please click here to view a larger version of this figure.

Figure 4: Needle preparation. (A) Micropipette puller. (B) Tip of a microinjection needle after pulling by a micropipette puller. The dotted box indicates the magnified needle tip. Scale bar represents 1 mm. Please click here to view a larger version of this figure.

Figure 5: Embryo microinjections. (A) The whole set of a microinjection system containing a microscope (middle) and an electronic microinjector (left) connected to a micromanipulator (right). (B) Embryos and microinjection needle. (C) The injection site of the embryo is labeled with the red arrow. Scale bar represents 200 µm. (D) A hatched larva under the microscope. Scale bar represents 1 mm. Please click here to view a larger version of this figure.
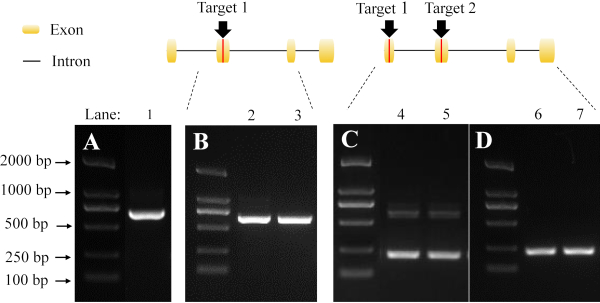
Figure 6: Detection of mutants by PCR and gel electrophoresis. The black arrows and red lines indicate the target sites of the sgRNA. (A) The band in lane 1 represents the amplification fragment derived from wild type. (B) The bands in lane 2 and 3 represent the amplification fragment derived from mutant using a single sgRNA target. (C) The detection of a heterozygote using a pair of non-overlapping sgRNA. The bands in lanes 4 and 5 represent the amplification fragment derived from the mutation of two sgRNA targets. The lower bands indicate a large fragment deletion. (D) The results are derived from a homozygote. The bands in lane 6 and 7 indicate the large fragment deletion. Please click here to view a larger version of this figure.
