The impact of fine-tuning reaction compounds on the final yield or quality of synthesized MPs is exemplified. A frequent routine approach is to adjust the optimal Mg2+ concentration in CF reactions by expression of green fluorescent protein (GFP) as a convenient monitor of system efficiency. As an example, GFP was synthesized from a pET-21a(+) vector at Mg2+ concentrations between 14 and 24 mM (Figure 2). SDS-PAGE analysis identified the optimal Mg2+ concentration at 20 mM (Figure 2A), which is in good accordance with complementary fluorescence measurements (excitation at 485 nm, emission measurement at 535 nm) of the CF reaction supernatant (Figure 2B). For the CF expression of the bacterial PR expressed from pIVEX 2.3 vector and of the turkey β1 adrenergic receptor (Tβ1AR) expressed from pET-21a(+) vector, the optimal Mg2+ concentration was identified at 20-22 mM.
As a further example, quality refinement of synthesized MPs with the described strategy is shown. Tunable parameters for the co-translational insertion of MPs into preformed nanodiscs are (i) the lipid composition of the nanodisc membrane and (ii) the final nanodisc concentration in the reaction. It is well known that the hydrophobic environment is an important parameter for correct folding, oligomeric assembly, and stability of MPs. Since lipid composition in nanodiscs can be modulated, the described approach enables a straightforward systematic screening of lipid effects on structure and function of a MP. In initial experiments, it is recommended to screen lipids with phosphatidylcholine (PC) and phosphatidylglycerol (PG) headgroups and to test different chain lengths and saturations. The impact of membrane composition and nanodisc concentration on the solubilization efficiency and quality of two different MPs is shown. PR is a light activated proton pump and a very stable and highly synthesized MP reaching final concentrations of > 100 µM in the RM. Thus, it is recommended to use it as a positive control to ensure correct reaction setup as PR concentration can be easily assessed by measurement of absorption at 530 nm in the RM supernatant. In contrast, Tβ1AR is an example of the large family of eukaryotic G-protein coupled receptors (GPCRs) and is a complex and unstable MP. For convenient monitoring, a Tβ1AR-GFP fusion construct was synthesized and the total concentration of nanodisc solubilized receptor in the CF reactions was determined via GFP fluorescence (Figure 3A). The fraction of functionally folded and ligand-binding active receptor was further determined using a filter binding assay and the labeled ligand [3H]-dihydroalprenolol (Figure 3B). The filter binding assay was performed as described previously14. Briefly, Tβ1AR-GFP concentration in the RM was determined via its fluorescence. The GPCR was incubated with the radiolabelled ligand for 1 hour at 20 °C, applied to a filter, and unbound ligand was washed off. For determination of unspecific [3H]-dihydroalprenolol binding, the receptor was saturated with unlabeled alprenolol in a control reaction. The counts of the radioactive ligand in the filters were measured in a scintillation counter and the amount of bound ligand was determined via its specific label activity. Percent GPCR binding activity was then calculated from the amount of bound ligand, the Tβ1AR-GFP concentration, and the assay volume. The overall synthesis and nanodisc solubilization of Tβ1AR-GFP is similar with all analyzed membrane compositions and within the range of 8-13 µM (Figure 3A). In contrast, a much higher variation is detectable in the quality of the synthesized GPCR. Lowest activity with less than 10% active fraction is obtained with the lipids DMPC and POPC. With DOPG and POPG, the active fraction of Tβ1AR-GFP could be increased to approximately 30% (Figure 3B). The results indicate that charge of the lipid headgroup as well as flexibility of the fatty acid chain are important modulators for folding and activity of this GPCR.
Besides nanodisc composition, their final concentration in the CF reaction can be an important factor for MP quality. Once a suitable membrane composition of a nanodisc has been identified, its concentration during MP synthesis should be screened. It is obvious that the nanodisc concentration needs to be adjusted according to the expression efficiency of a MP. CF synthesis of Tβ1AR-GFP receptor and PR result in final concentrations of approximately 10 µM and 100 µM in the RM, respectively. If the nanodisc concentration is screened within a range of 3.75-60 µM, a complete solubilization of the GPCR is obtained at approximately 30 µM nanodiscs, giving a ratio of Tβ1AR:nanodisc of 1:3 (Figure 4A). In contrast, complete solubilization of PR is already achieved with approximately 10 µM nanodiscs and gives a PR:nanodisc ratio of 10:1 (Figure 4B). An excess of nanodiscs is therefore necessary to achieve almost complete solubilization of Tβ1AR-GFP, while an inverted ratio in the case of PR indicates the insertion of multiple PR copies into one nanodisc. Subsequent studies of purified PR/nanodisc complexes with native mass spectrometry confirmed this observation and found a prevalence of higher oligomeric forms of PR if synthesized at lower nanodisc concentrations15,18. The titration of CF synthesized MPs with nanodiscs can therefore be used as a tool to trigger oligomeric assemblies and to study their effects on MP function.
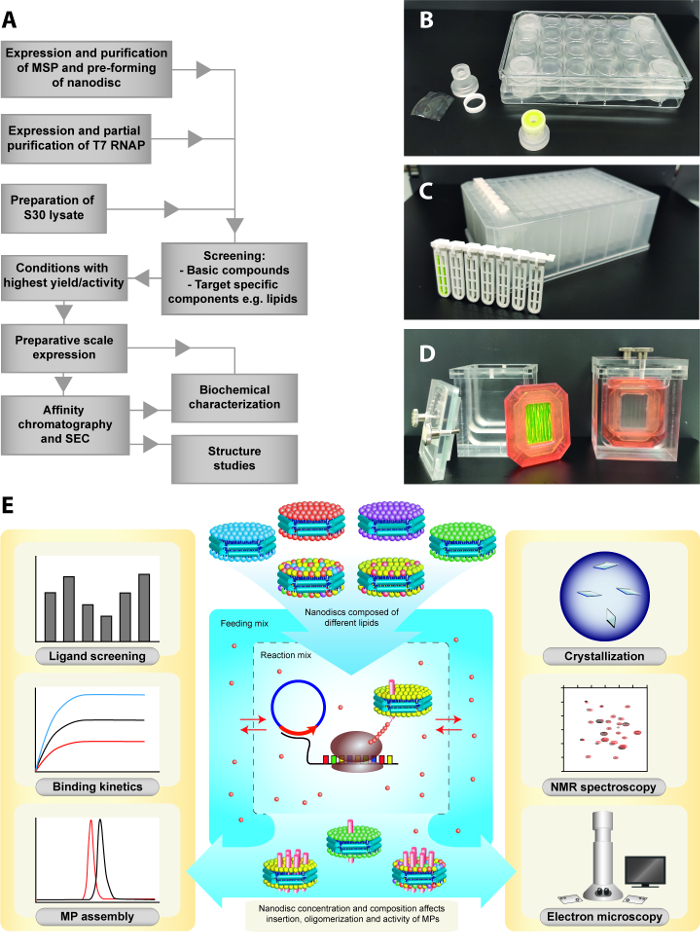
Figure 1: CECF expression strategy for the insertion of membrane proteins into nanodiscs. (A) Basic workflow illustrating the major steps of the process. (B) Customized analytical scale reaction vessels for CECF expression. (C) Commercially available dialysis cartridges for analytical scale CECF setup. (D) Preparative scale setup (3 mL RM) including a 3 mL dialysis cassette and a customized plexiglass FM container. (E) Co-translational insertion of MPs into preformed nanodiscs and lipid screening in the CECF configuration. The RM contains the required transcription/translation machinery and nanodiscs while low molecular weight compounds are present in both compartments. Biochemical and structural applications of the produced MP/nanodisc samples are further illustrated. Please click here to view a larger version of this figure.
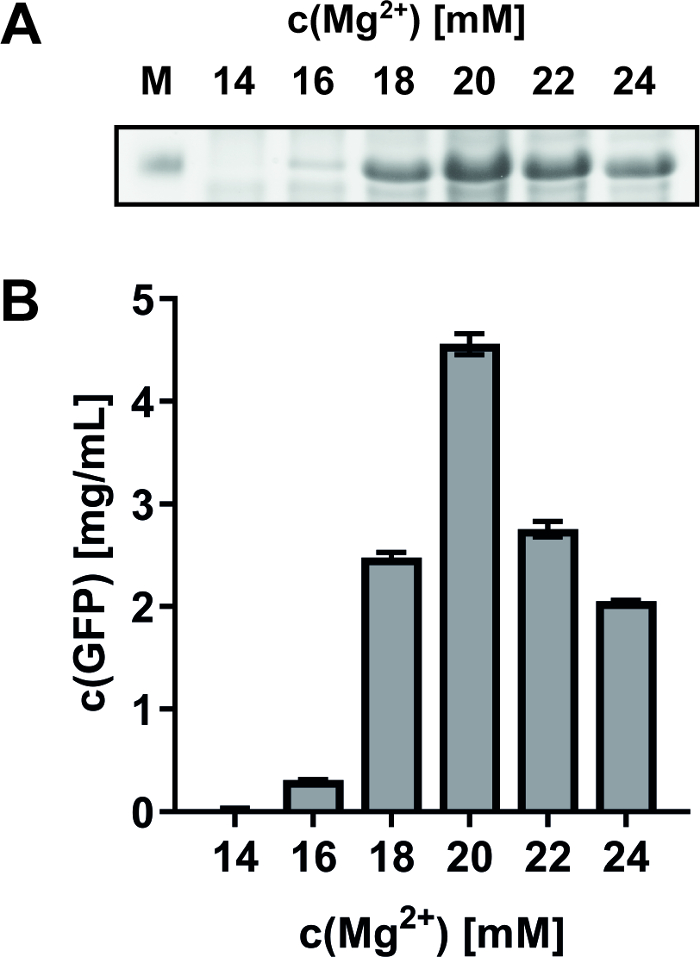
Figure 2: Effect of different Mg2+ concentrations on CF GFP synthesis. GFP was synthesized in CF reactions with Mg2+ concentrations within a range of 14-24 mM. (A) SDS-PAGE analysis shows the strongest band of GFP at 20 mM Mg2+. M: Marker. (B) GFP fluorescence after CF expression with different Mg2+ concentrations. The maximal GFP fluorescence at 20 mM Mg2+ corresponds to 4.6 mg/mL. Error bars represent the standard deviation of a duplicate measurement. Please click here to view a larger version of this figure.
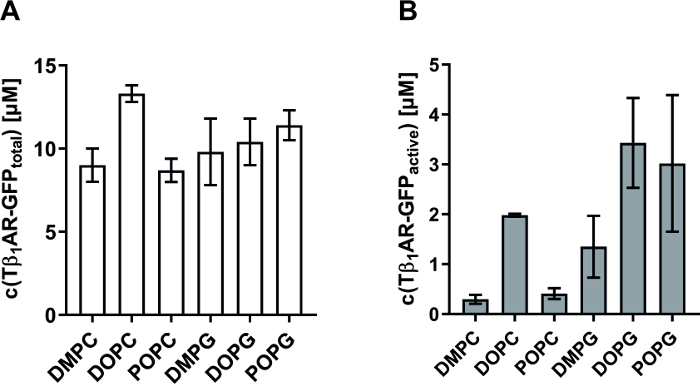
Figure 3: Effect of nanodisc membrane composition on solubilization and quality of a CF synthesized GPCR. Yield and activity of Tβ1AR-GFP synthesized in the presence of 30 µM nanodiscs containing different membrane compositions. The total synthesized protein (A) and fraction of ligand-binding active receptor (B) are given in µM. Total concentration was determined via fluorescence measurement of the GFP fusion. Activity was measured via filter binding assay with the radiolabeled ligand [3H]-dihydroalprenolol. Error bars represent standard deviations of independent triplicates. Data taken from previously published manuscript14. Please click here to view a larger version of this figure.
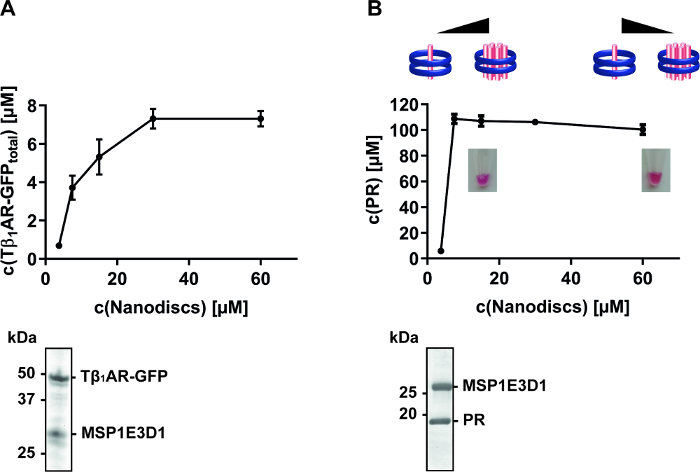
Figure 4: Solubilization screen of CF synthesized MPs with increasing nanodisc concentrations. The MPs were synthesized in the presence of supplied nanodiscs within a range of 3.75-60 µM. (A) Expression of Tβ1AR-GFP with nanodiscs (DMPC) in the CF reaction. Total concentration was determined via fluorescence measurement of the GFP fusion. Data taken from previously published manuscript14. Affinity-tag purified samples were analyzed by SDS-PAGE and Coomassie-Blue staining shows two prominent bands corresponding to Tβ1AR-GFP at approximately 50 kDa and MSP1E3D1 above 25 kDa (B) Expression of PR with nanodiscs (DOPG) in the CF reaction. Total concentration of PR was determined via absorption measurement at 530 nm. Data taken from previously published manuscript10,18. The photos show the red color of the final reactions due to the presence of folded PR. The pictograms illustrate the modulated PR assembly towards lower oligomeric conformations upon increased nanodisc concentrations, as revealed by subsequent native mass spectrometry18. Affinity-tag purified samples were analyzed by SDS-PAGE and Coomassie-Blue staining shows two prominent bands corresponding to PR at approximately 20 kDa and MSP1E3D1 above 25 kDa. Please click here to view a larger version of this figure.
| Compound | Final concentration range | Effect on MP sample |
| Mg2+ | 10 – 30 mM | yield |
| DTT | 1 – 20 mM | disulfide bridge formation, folding |
| 20 Amino acid mixture1 | 0.2 – 2 mM | yield, |
| GSSG : GSH2 | (1-10 µM) : (1-10 µM) | disulfide bridge formation, folding |
| Nanodiscs | 10 – 100 µM | solubilization, oligomeric assembly, folding |
| Lipid type | solubilization, oligomeric assembly, folding | |
| S30 : HS30 lysate3 | (50-100 %) : (50-100 %) | folding |
| S12 – S100 | 20 – 50 % | yield, MP background in channel assays |
| DNA template | 0.5 – 30 ng/µL RM | yield, oligomeric assembly, folding |
| DNA template design | yield | |
| 1, mixture may be varied according to the individual composition of a MP to e. g. improve yield or optimize NMR labelling schemes. 2, GSH should always be prepared freshly. 3, total CF lysate concentration in the RM should be at least 35 % with a S30 content of at least 50 % in order to ensure high expression levels. |
||
Table 1: Critical screening components of CF reactions.
| MSP1E3D1 | Lipid | DPC | ||||||
| Ratio | (410 µM stock) | (50 000 µM stocks) | (10 % (w/v) stock) | |||||
| Lipid | Lipid : MSP | V(MSP) | c(MSPfinal) | V(lipid) | c(lipidfinal) | V(DPC) | c(DPCfinal) | DF buffer |
| [µL] | [µM] | [µL] | [µM] | [µL] | [%] | [µL] | ||
| DMPC | 115 : 1 | 1500.0 | 205.0 | 1415.0 | 23 575 | 30.0 | 0.1 | 55.5 |
| DOPC | 80 :1 | 1500.0 | 205.0 | 984.0 | 16 400 | 30.0 | 0.1 | 486.0 |
| POPC | 85 : 1 | 1500.0 | 205.0 | 1046.0 | 17 425 | 30.0 | 0.1 | 425.0 |
| DMPG | 110 : 1 | 1500.0 | 205.0 | 1353.0 | 22 550 | 30.0 | 0.1 | 117.0 |
| DOPG | 80 : 1 | 1500.0 | 205.0 | 984.0 | 16 400 | 30.0 | 0.1 | 486.0 |
| POPG | 90 : 1 | 1500.0 | 205.0 | 1107.0 | 18 450 | 30.0 | 0.1 | 363.0 |
Table 2: Lipid to MSP1E3D1 ratios for 3 mL in vitro assembly setups.
| Compound | Concentration | Preparation |
| DNA plasmid template1 | > 400 µg/mL in 10 mM Tris-HCl pH 8.0 | Midi/Maxi kit (e.g. Qiagen) preparation2 |
| Mixture of 20 amino acids3 | 25 mM each in H2O | precipitate remains4 |
| Mixture of 4 NTPs (75 x) | c(CTP) 240 mM, c(ATP) 360 mM, c(UTP) 240 mM, c(GTP) 240 mM in H2O. Adjust pH to 7-8 with KOH |
A little precipitate remains4 |
| Acetyl phosphate | 1 M in H2O, adjust pH 7-8 with KOH | precipitate remains4 |
| Phospho(enol)pyruvic acid (K+) | 1 M in H2O, adjust pH 7-8 with KOH | |
| Folinic acid | 10 mg/mL in H2O | precipitate remains4 |
| DTT | 0.5 M in H2O | |
| c0mplete protease inhibitor cocktail | 1 tablet per 1 mL in H2O | |
| Tris-acetate, pH 8.0 | 2.4 M in H2O | |
| Mg(OAc)2 | 1 M in H2O | |
| KOAc | 10 M in H2O | |
| Ribolock RNase-Inhibitor | 40 U/mL | Thermo Fisher Scientific |
| tRNA (E. coli) | 40 mg/mL in H2O | Roche (Germany) |
| T7RNAP | 3-7 mg/mL5 | see protocol section 2 |
| Pyruvate kinase | 10 mg/mL | Roche (Germany) |
| Nanodiscs (DMPG) | 0.2-1.0 mM6 in 10 mM Tris-Cl, pH 8.0, 100 mM NaCl | see protocol sections 3 and 4 |
| 1, PCR template could be used at similar concentrations. 2, the quality of “Mini”-kit prepared DNA is not satisfactory. 3, cysteine tends to be unstable and may be added separately. 4, solution is oversaturated, thorough mixing instantly before removing aliquots is necessary. 5, for each new T7RNAP batch, an initial screen is recommended to identify the best final concentration. 6, solubility of nanodiscs can depend on their lipid composition. |
||
Table 3: Preparation of CF stock solutions.
| Compound | ||
| For Mastermix (3.0 x) | c(final) | V [µL] |
| Mixture of 20 amino acids | 1 mM | 2520.0 |
| Mixture of 4 NTPs (75 x) | 1x | 840.0 |
| Acetyl phosphate | 20 mM | 1260.0 |
| Phospho(enol) pyruvic acid |
20 mM | 1260.0 |
| Folinic acid | 0.1 mg/mL | 630.0 |
| Tris-acetate, pH 8.0 | 100 mM | 2625.0 |
| c0mplete 50 x | 1x | 1260.0 |
| Mg(OAc)2 | 19.8 (= 20)1 mM | 1260.0 |
| KOAc | 180 (= 270)2 mM | 1140.0 |
| DTT | 2 mM | 252.0 |
| H2O | 7953.0 | |
| Total | 21 000 | |
| For RM | c(final) | V [µL] |
| 3x Mastermix | 1 x | 1000.0 |
| RNase inhibitor | 0.3 U/µL | 22.5 |
| tRNA (E. coli) | 0.5 mg/mL | 37.5 |
| Nanodiscs (DMPG)3 | 10 µM | 75.0 |
| DNA template4 | 0.015 ng/µL | 112.5 |
| Pyruvate kinase | 0.04 mg/mL | 12.0 |
| T7RNAP5 | 0.03 mg/mL | 15.0 |
| S30 lysate | 0.35 % | 1050.0 |
| All-trans retinal6 | 0.6 mM | 9.0 |
| H2O | – | 666.5 |
| Total | 3000.0 | |
| For FM | c(final) | V [µL] |
| Mastermix (3 x) | 1 x | 20 000 |
| H2O | – | 40 000 |
| Total | 60 000 | |
| 1, 0.2 mM Mg2+ are contributed from the S30 lysate. 2, 90 mM K+ are contributed from acetyl phosphate and phospho(enol) pyruvic acid. 3, calculated stock solution is 400 µM. 4, calculated stock solution is 400 µg/mL. 5, calculated stock solution is 6 mg/mL. 6, specific cofactor for PR, stock solution is 200 mM in DMSO. |
||
Table 4: Pipetting scheme for a CECF reaction with 3 mL of RM and 60 mL of FM
| Compound | ||||||||
| For Mastermix (3.0 x) | c(final) | V [µL] | ||||||
| Mixture of 20 amino acids | 1 mM | 864.0 | ||||||
| Mixture of 4 NTPs | 1x | 288.0 | ||||||
| Acetyl phosphate | 20 mM | 432.0 | ||||||
| Phospho(enol)pyruvic acid | 20 mM | 432.0 | ||||||
| Folinic acid | 0.1 mg/mL | 216.0 | ||||||
| Tris-acetate, pH 8.0 | 100 mM | 900.0 | ||||||
| c0mplete | 1x | 432.0 | ||||||
| Mg(OAc)2 | 13,8 (= 14) mM1 | 298.0 | ||||||
| KOAc | 180 (= 270) mM2 | 389.0 | ||||||
| DTT | 2 mM | 86.4 | ||||||
| H2O | – | 2862.6 | ||||||
| Total [µL]1 | – | 7200.0 | ||||||
| Mg2+ concentration | ||||||||
| 14 mM | 16 mM | 18 mM | 20 mM | 22 mM | 24 mM | |||
| For RM | c(final) | V [µL] | V [µL] | V [µL] | V [µL] | V [µL] | V [µL] | |
| 3x Mastermix | 1 x | 67.0 | 67.0 | 67.0 | 67.0 | 67.0 | 67.0 | |
| 0.1 M Mg(OAc)2 | 14-24 mM | 0.0 | 4.0 | 8.0 | 12.0 | 16.0 | 20.0 | |
| RNase inhibitor | 0.3 U/µL | 1.5 | 1.5 | 1.5 | 1.5 | 1.5 | 1.5 | |
| tRNA (E. coli) | 0.5 mg/mL | 2.5 | 2.5 | 2.5 | 2.5 | 2.5 | 2.5 | |
| DNA template3 | 0.015 ng/µL | 7.5 | 7.5 | 7.5 | 7.5 | 7.5 | 7.5 | |
| Pyruvate kinase | 0.04 mg/mL | 0.8 | 0.8 | 0.8 | 0.8 | 0.8 | 0.8 | |
| T7RNAP4 | 0.03 mg/mL | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | |
| S30 lysate | 35 % | 70.0 | 70.0 | 70.0 | 70.0 | 70.0 | 70.0 | |
| H2O | – | 49.7 | 45.7 | 41.7 | 37.7 | 33.7 | 29.7 | |
| Total [µL]5 | – | 200.0 | 200.0 | 200.0 | 200.0 | 200.0 | 200.0 | |
| Mg2+ concentration | ||||||||
| 14 mM | 16 mM | 18 mM | 20 mM | 22 mM | 24 mM | |||
| For RM | c(final) | V [µL] | V [µL] | V [µL] | V [µL] | V [µL] | V [µL] | |
| 3x Mastermix | 1 x | 1133.0 | 1133.0 | 1133.0 | 1133.0 | 1133.0 | 1133.0 | |
| 0.1 M Mg(OAc)2 | 14-24 mM | 0.0 | 68.0 | 136.0 | 204.0 | 272.0 | 340.0 | |
| H2O | – | 2267.0 | 2199.0 | 2131.0 | 2064.0 | 1995.0 | 1927.0 | |
| Total [µL]5 | – | 3400.0 | 3400.0 | 3400.0 | 3400.0 | 3400.0 | 3400.0 | |
| 1, 0.2 mM Mg2+ are contributed from the S30 lysate. The mastermix is adjusted to the lowest screening concentration. 2, 90 mM K+ are contributed from acetly phosphate and phospho(enol) pyruvic acid. 3, calculated stock solution is 400 µg/mL. 4, calculated stock solution is 6 mg/mL. 5, reactions are performed in duplicates. |
||||||||
Table 5: Pipetting scheme for Mg2+ concentration screen with 100 µL of RM and 1.7 mL of FM.
