For beads preparation, we determined the optimal amount of Rhodamine Red-X succinimidyl ester for the transgenic strain expressing GFP-myosin II and mCherry-histone (Figure 1A-D). We used mCherry tagged histone as a marker of cell cycle progression. Because both Rhodamine Red-X and mCherry will be illuminated by a 561 nm laser, the optimal intensity of Rhodamine Red-X signal is comparable to that of histone to allow simultaneous imaging of cell and bead. For example, the fluorescence signal from the bead treated with 0.005 µg/mL Rhodamine Red-X succinimidyl ester was too weak to visualize the bead (Figure 1A). On the other hand, the fluorescence signal from the beads treated with 5 µg/mL Rhodamine Red-X succinimidyl ester was too strong to image mCherry-histone (Figure 1D). We determined that 0.5 µg/mL Rhodamine Red-X succinimidyl ester is optimal for this particular transgenic strain (Figure 1C).
Blastomere isolation was performed according to the procedures shown in Figure 4. The following points should be addressed for successful experiments. 1) The media in the well evaporates over time and becomes viscous; this causes the embryos to stick to the glass capillary and other embryos, so a new well with fresh media needs to be used. 2) In the hypochlorite solution, embryos float to the surface. Therefore, lower the magnification of the microscope and focus on the surface of the droplet to ease with the transferring of embryos. 3) The effectiveness of hypochlorite solution may differ. Hence, duration of embryos spend in the hypochlorite solution should be tested for each new batch of hypochlorite solution made. 4) Place the mouth pipette as close to the embryo as possible to minimize the strength used to blow into the pipette, but also not too close so that the embryo is sucked up as well (this is also true when attaching beads). 5) After eggshell removal, embryos become very delicate. Hence, forces exerted on the mouth pipette need to be slightly lowered to prevent embryos from bursting. 6) Prolonged usage of the mouth pipette may dampen the PTFE filter, and 7) 2-cell stage embryos should only be separated when the nucleus has been clearly formed (Figure 4C, left schematics). Compared to cells in intact embryos (Figure 4A; right), cells in embryos without eggshell look rounder (Figure 4C; right). Additionally, after blastomere isolation, the shape of blastomeres become spherical (Figure 4D; right).
To test the effects of physical contact-dependent cue on cell division orientation, we attached Rhodamine Red-X coated beads to AB blastomeres isolated from the 2-cell stage and performed live-imaging (Figure 6, Figure 7). The bead without Rhodamine Red-X treatment did not adhere to the blastomeres, suggesting that the Rhodamine Red-X serves as a both fluorescence marker and adhesive molecule. For the analysis of cell division orientation, we have 3D reconstructed the time-lapse image using the "3D project" function of ImageJ12 (Figure 6A; tilting around Y-axis). To determine the plane containing mitotic spindle (spindle plane), we first identified a plane where two centrosomes were vertically aligned (Figure 6B; 110°): the plane with ± 90° angle of this plane is spindle plane (Figure 6B; -20°). Next, we have measured the angle of spindle (spindle orientation) relative to the cell-bead contact interface (contact orientation) after cytokinesis (Figure 6C). When both daughter cells were attached to the bead, a line that passes both contact sites was used as a contact orientation.
In our previous study, we have identified an anisotropic cell surface myosin flow during the bead-induced AB cell division orientation10. However, it is difficult to measure the intracellular myosin flow as cell moves during oriented division. To perform this, first we have selected the samples with spindle aligned to the imaging plane (xy plane) (Figure 7). Second, Z-stacks were projected using maximum projection method (Figure 7). Third, cell movements were corrected using the subpixel registration algorithm Stack reg plug-in13 of ImageJ with Rigid body option (Figure 7B). By the registration of images, the position of cell was stabled (Figure 7B). Finally, by using Manual tracking plug-in of ImageJ, myosin foci were tracked (Figure 7C). According to the coordinate information, velocities of myosin along division axis, and axis perpendicular to it were calculated within 50 s after the cytokinesis onset.
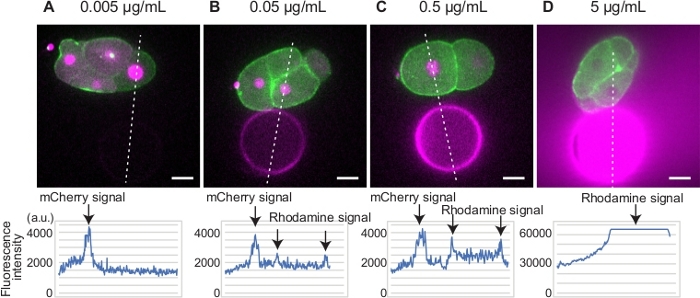
Figure 1: Determination of Rhodamine Red-X concentration. (A-D) Embryos expressing GFP-myosin (green) and mCherry-histone (magenta) were placed in proximity to beads bound with different concentrations of Rhodamine Red-X (magenta). The signal intensities of mCherry and Rhodamine Red-X are plotted along the white dotted lines (bottom graphs). The beads and histone signals were clearly detected for beads treated with 0.5 µg/mL Rhodamine Red-X. Scale bars show 10 µm. Please click here to view a larger version of this figure.

Figure 2: Assembly of mouth pipette. Mouth pipette is assembled by connecting narrow (1) and wide (2) Tygon tubes with a PTFE filter (3). At the end of the narrow and wide Tygon tube, capillary holder (4) and mouth piece (5) were attached, respectively. A hand-drawn glass capillary (6) can be inserted in the capillary holder. Scale bar is 50 mm. Please click here to view a larger version of this figure.
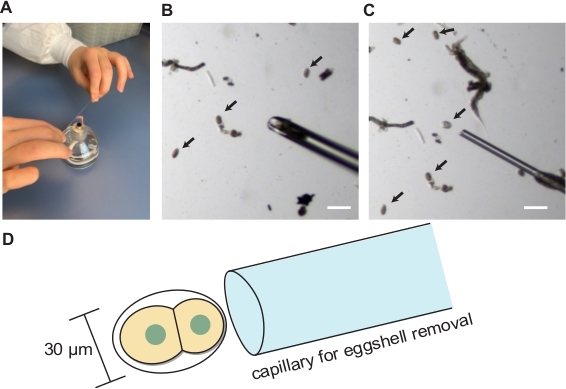
Figure 3: Blastomere isolation. (A) Hand pulling of glass capillary. (B) Hand-drawn glass capillary for embryo transfer. (C) Hand-drawn glass capillary for eggshell removal. (D) Schematics showing the appropriate size of capillary opening for the eggshell removal. Arrows indicate embryos. Scale bars show 100 µm. Please click here to view a larger version of this figure.
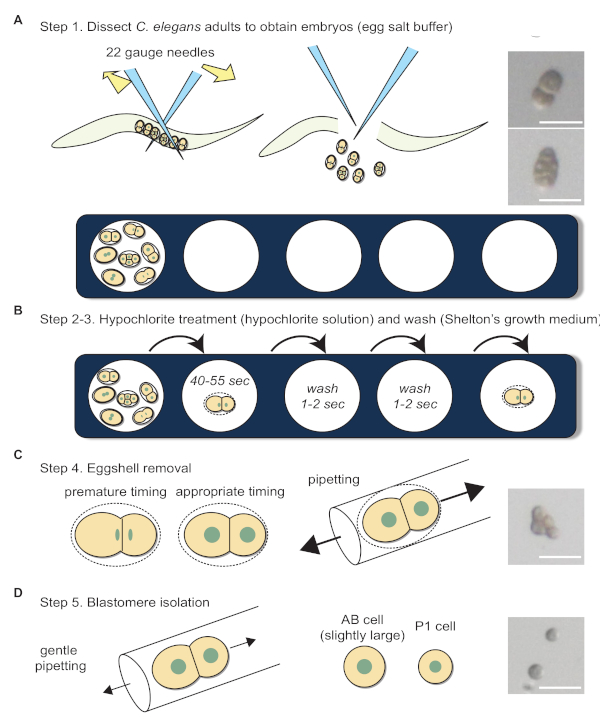
Figure 4: Blastomere isolation workflow. (A) Dissection of adult C. elegans in egg salt buffer to obtain embryos. Photographs show 2-cell and 4-cell stage embryos before eggshell removal. (B) Hypochlorite treatment and washing. (C) Schematics depict the appropriate timing for eggshell removal. Photograph shows a 4-cell stage embryo after eggshell removal. (D) Blastomere separation. Photograph shows a separated 2-cell stage embryo. Sizes of the arrows in C and D indicate the relative forces required during pipetting. Scale bars show 50 µm. Please click here to view a larger version of this figure.
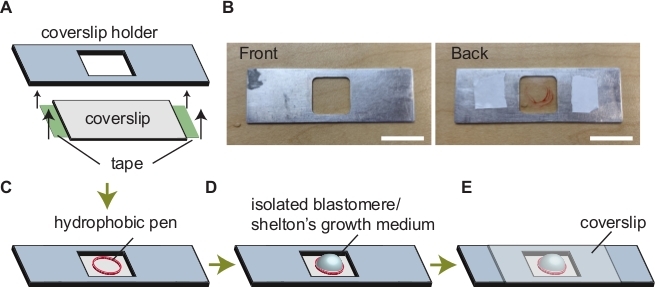
Figure 5: Assembly of imaging chamber. (A) Attaching coverslip to the coverslip holder. (B) Images of a coverslip holder. Coverslip holder before and after assembly is indicated on the left and right, respectively. (C) A circle drawn via a hydrophobic pen. (D) Addition of Shelton's growth medium and sample within the circle. (E) Mounting a coverslip to avoid evaporation. Scale bars show 50 mm. Please click here to view a larger version of this figure.
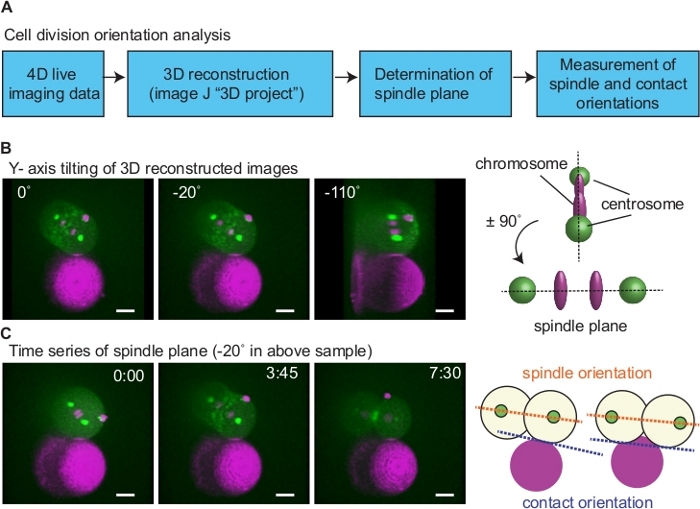
Figure 6: Analysis of cell division orientation. (A) A diagram of cell division orientation analysis. (B) Determination of spindle plane. Left images show an example of a sample. 3D reconstructed 4-D movies were rotated around Y-axis to determine the plane wherein two centrosomes align vertically (right image; right upper schematics). In this example, spindle plane is ± 90° of 110° (middle image; right bottom schematics). (C) Measurement of spindle orientation relative to the cell-bead contact. Using images of the spindle plane, spindle orientation after cytokinesis was determined based on angle between lines connecting two centrosomes (orange dotted lines in the right schematics) and cell contact (blue dotted lines). When both daughter cells were attached to the beads, cell-bead contact orientation was the line that passes both contact sites. Green is myosin and centrosome, magenta is histone and beads. Scale bars are 10 µm. Times are minutes and seconds. Please click here to view a larger version of this figure.
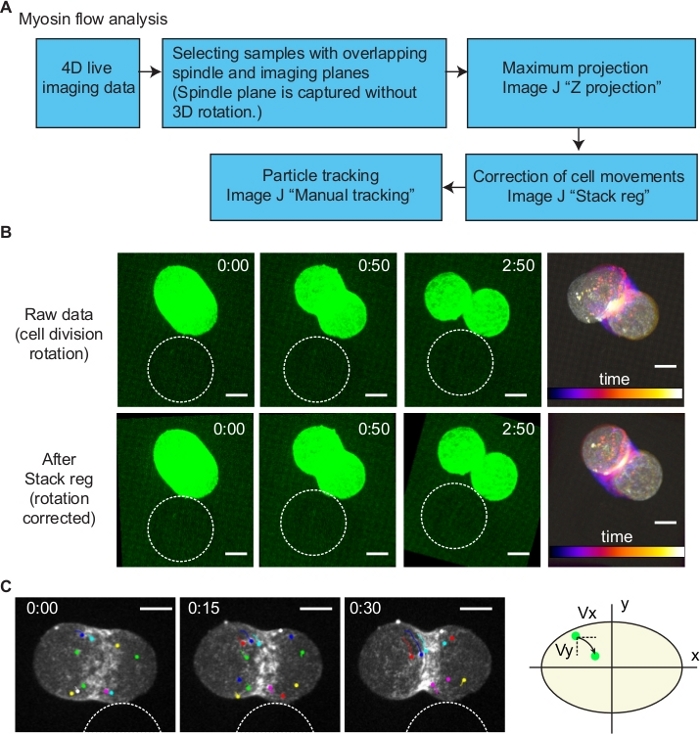
Figure 7: Analysis of myosin flow. (A) A diagram of myosin flow analysis. (B) Correction of cell division orientation. To quantify intracellular myosin flow dynamics, the rotation of dividing cell was corrected using the ImageJ plugin Stack reg (Rigid body option). Upper and bottom images are before and after the Stack reg processing. Right most images show temporal color code of time series. (C) Tracking of myosin foci. Using the images processed by Stack reg, individual myosin foci movements were tracked by ImageJ Manual tracking plugin. Division axis and the axis perpendicular to it were defined as x and y, respectively (right schematics). Myosin flow velocities in x and y axis (Vx and Vy) were measured according to the coordinate information. Scale bars are 10 µm. Times are minutes and seconds. Please click here to view a larger version of this figure.
