Schematic of steps involved in library preparation
An overall schematic of small RNA extraction, sequencing, and alignment is outlined in Figure 2.
Liver samples from one male and one female mouse were collected and snap frozen in liquid nitrogen. Total RNA was extracted and evaluated for quality and concentration.
Small RNA sequencing yields sufficient RNA for sequencing
3 μg of RNA from two independent RNA extractions were used as starting material for small RNA sequencing. Samples were run on an acrylamide gel and cut out between size markers corresponding to 17-28 nt of RNA (Figure 1A). Samples were chopped into fragments for RNA isolation (Figure 1B) and transferred to a low-retention 1.5 mL centrifuge tube (Figure 1C). Barcodes bc7 and bc17 (Table 1) were ligated to the 5' end of the small RNA. Small RNA libraries were PCR amplified using 22 cycles of PCR to yield 8.0 and 11.2 ng/μL product, respectively. Samples were pooled and a 10 nM pooled sample was submitted for high-throughput sequencing using a 50 bp read length.
MiR-122 is the most abundant microRNA in the mouse liver
After barcode sorting, 851,931 reads contained barcodes from liver sample 1 and 650,154 from liver sample 2. Of the reads, 83.5% and 90.0% mapped to microRNAs respectively, with the remaining reads mapping to rRNAs (1.8% and 0.6% respectively), tRNAs and mRNA degradation fragments. After alignment to human microRNA hairpins, we observed strong concordance between microRNA read counts in each replicate (R2 = 0.998; Figure 3). A total of 306 microRNA species were detected, with the greatest number of reads mapping to miR-122 (Supplementary Table 4). MicroRNA abundance was similar between male and female liver samples.
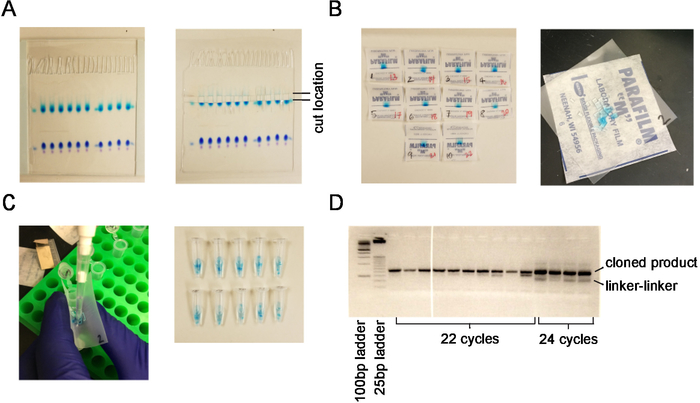
Figure 1. Extraction of small RNAs from an acrylamide gel. (A) Acrylamide gel and region that is cut corresponding to the size of microRNAs. (B) Gel pieces before and after cutting into smaller fragments. (C) Process for transferring gel fragments into siliconized tubes. (D) PCR reaction on low-melt agarose gel demonstrating correct cloned product compared with linker-linker product and unsaturated (22 cycles) versus saturated (24 cycles) samples. Please click here to view a larger version of this figure.
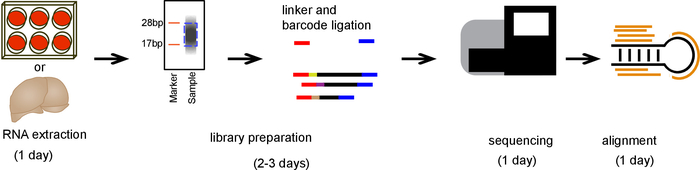
Figure 2. Schematic of protocol. A timeline showing the major steps involved in the procedure. Please click here to view a larger version of this figure.
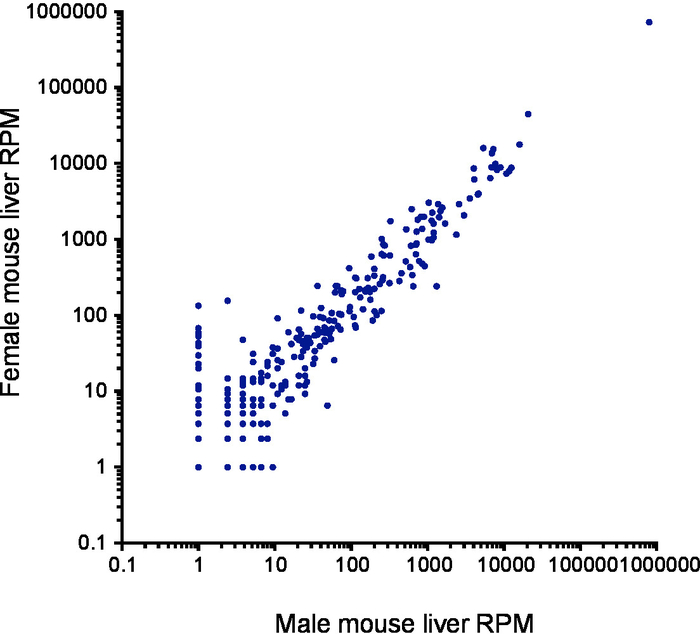
Figure 3. Reproducibility of results from two independent RNA extractions. Scatterplot of microRNA read counts from a male mouse liver (x-axis) compared with a female mouse liver (y-axis) using a log10-based scale. Each point represents the reads per million (RPM) mapped microRNA count for each individual microRNA. Please click here to view a larger version of this figure.
| Primer | Sequence | |
| 3' linker 1 | rAppCTGTAGGCACCATCAAT–NH2 | |
| lower size marker | rArUrCrGrCrArUrGrCrUrGrArCrGrUrArCrUrArGGTAACCGCATCATGCGTC | |
| upper size marker | rArArUrCrArGrCrGrGrArUrUrGrCrArUrGrArArCrGrUrArCrArUrArGGTAACCGCATCATGCGTC | |
| barcode1 | /5AmMC6/ACGCTCTTCCGATCTrArGrCrG | |
| barcode2 | /5AmMC6/ACGCTCTTCCGATCTrCrGrUrC | |
| barcode3 | /5AmMC6/ACGCTCTTCCGATCTrCrUrGrG | |
| barcode4 | /5AmMC6/ACGCTCTTCCGATCTrArCrUrU | |
| barcode5 | /5AmMC6/ACGCTCTTCCGATCTrGrGrGrU | |
| barcode6 | /5AmMC6/ACGCTCTTCCGATCTrGrUrUrA | |
| barcode7 | /5AmMC6/ACGCTCTTCCGATCTrUrArUrG | |
| barcode8 | /5AmMC6/ACGCTCTTCCGATCTrUrCrGrC | |
| barcode9 | /5AmMC6/ACGCTCTTCCGATCTrGrCrArG | |
| barcode10 | /5AmMC6/ACGCTCTTCCGATCTrArUrArC | |
| barcode11 | /5AmMC6/ACGCTCTTCCGATCTrUrUrCrU | |
| barcode12 | /5AmMC6/ACGCTCTTCCGATCTrCrArArU | |
| barcode13 | /5AmMC6/ACGCTCTTCCGATCTrArArGrA | |
| barcode14 | /5AmMC6/ACGCTCTTCCGATCTrUrGrArA | |
| barcode15 | /5AmMC6/ACGCTCTTCCGATCTrUrGrGrG | |
| barcode16 | /5AmMC6/ACGCTCTTCCGATCTrArUrUrG | |
| barcode17 | /5AmMC6/ACGCTCTTCCGATCTrUrCrArU | |
| barcode18 | /5AmMC6/ACGCTCTTCCGATCTrGrUrArU | |
| RT primer | ATTGATGGTGCCTACAG | |
| PCR primer F | AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT | |
| PCR primer R | CAAGCAGAAGACGGCATACGAGCTCTTCCGATCTATTGATGGTGCCTACAG | |
Table 1. List of primers.
Supplemental Table 1. List of barcode sequences. Please click here to download this file.
Supplemental Table 2. Curated list of mouse microRNA precursor sequences. Please click here to download this file.
Supplemental Table 3. Curated list of human microRNA precursor sequences. Please click here to download this file.
Supplemental Table 4. Raw and normalized microRNA read counts. Please click here to download this file.
