Lck-RCaMP2 and OER-GCaMP6f were expressed in HeLa cells, and both signals were recorded simultaneously using image-splitting optics, 24 h after transfection (Figure 3A and Video 1). The images were acquired at 10 Hz. Histamine (His, 1 µM), which induces Ca2+ release from the ER, was added during the recording. Upon the application of His, the signal intensity of Lck-RCaMP2 and OER-GCaMP6f increased, as shown by the pseudocolor display of ΔF/F0, which represents the change from the initial fluorescence intensity (Figure 3B). The time courses of Ca2+ elevation (ΔF/F0) reported by Lck-RCaMP2 and OER-GCaMP6f were compared in the same region of interest (ROI) (Figure 3C). The ΔF/F0 values were normalized to their peak values to enable the time course comparison between the two different GECIs, which have different expression levels and distributions. Both sensors reported an oscillation-like Ca2+ elevation. Lck-RCaMP2 and OER-GCaMP6f showed the same time course for Ca2+ elevation in two cells among the five cell types examined (Figure 3C, ROI 1 and 3). However, Ca2+ elevations shown by Lck-RCaMP2 remained at a higher level compared to that shown by OER-GCaMP6f (Figure 3C, ROI 2, 4, and 5). The results indicate that the Ca2+ elevation is prolonged in the vicinity of the plasma membrane, while it is terminated earlier around the ER, which is the source of this Ca2+ signal induced by His stimulation.
Spontaneous Ca2+ signals from astrocytes in the neuron-astrocyte mixed culture from rat hippocampi (Figure 4A) and cortexes (Figure 4B) were shown by Lck-RCaMP2 and OER-GCaMP6f (Figure 4, Video 2, Video 3, Video 4, and Video 5). Cortical cultures were revived from the frozen stock that was prepared as described in this protocol. Lck-RCaMP2 and OER-GCaMP6f signals were sequentially recorded at 2 Hz from the same cells. Three ROIs were selected in the area that showed Ca2+ elevation by each GECI, and the time course of ΔF/Fbase (i.e., the fluorescence intensity) changed from the average fluorescence intensity during the entire recording period (Fbase). When the baseline fluorescence is stable and Ca2+ elevations are less frequent, Fbase becomes a useful baseline to detect Ca2+ elevation events. Spontaneous Ca2+ elevations were visible only at the astrocytic process, not at the cell body. This result is consistent with the previous reports on astrocytic spontaneous Ca2+ signals by other GECIs visualized in vitro13 and in vivo14. In both hippocampal and cortical astrocytes, Ca2+ elevations shown by Lck-RCaMP2 (top) were more frequent than those shown by OER-GCaMP6f. This result is consistent with our previous demonstration that Ca2+ elevations in astrocytes due to Lck-GCaMP6f were more frequently detected than those due to OER-GCaMP6f7 and suggests that this notion is also applicable at the single-cell level.
Spontaneous Ca2+ elevations by Lck-GCaMP6f in immature rat hippocampal neurons (10 DIV) were seen at 2 Hz (Figure 5A and Video 6). The time courses of ΔF/F0 in five different ROIs suggest that these Ca2+ elevations are locally confined to the subcellular domains. Figure 5B (Video 7) shows the Ca2+ responses in mature mouse hippocampal neurons (30 DIV) infected with OER-GCaMP6f-expression AAV vectors. Neurons were stimulated with 100 µM DHPG, which is the agonist for metabotropic glutamate receptors, inducing Ca2+ release. DHPG-induced Ca2+ release due to OER-GCaMP6f was detected.
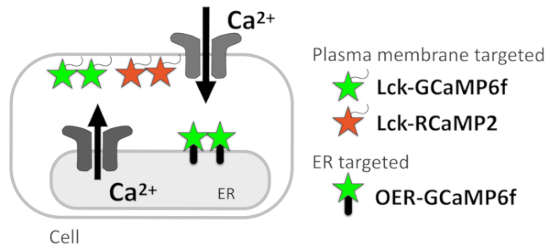
Figure 1: Diagram showing membrane-targeted GECIs. Schematic diagram of plasma membrane-targeted GECIs (Lck-GCaMP6f and Lck-RCaMP2) and outer ER membrane-targeted GCaMP6f (OER-GCaMP6f).
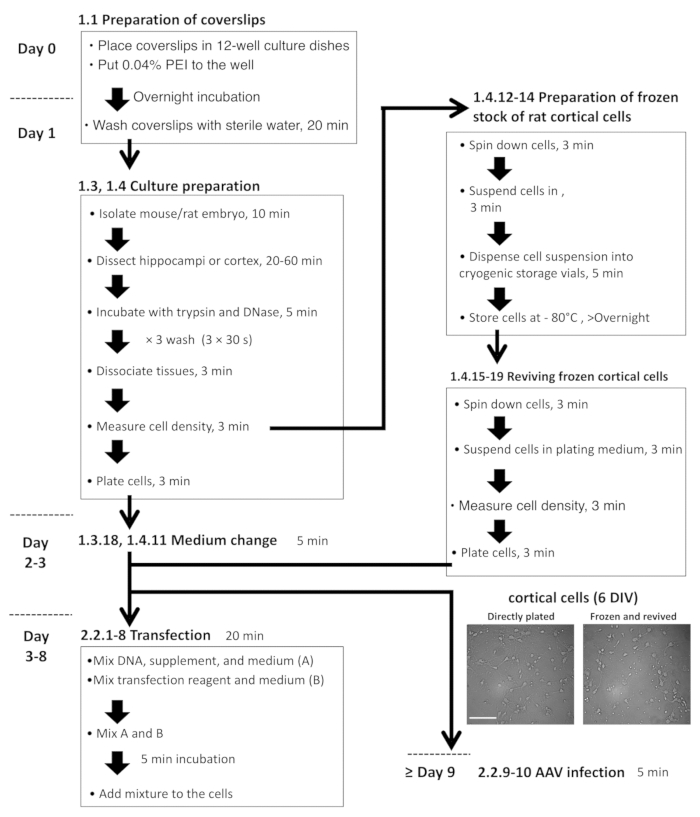
Figure 2: Flowchart for hippocampal and cortical cell preparation, plasmid transfection, and AAV infection. The microscopic images are representative, freshly plated DIV-6 cortical cells (left) and revived cells from the frozen stock (right). Scale bar = 100 µm. Please click here to view a larger version of this figure.
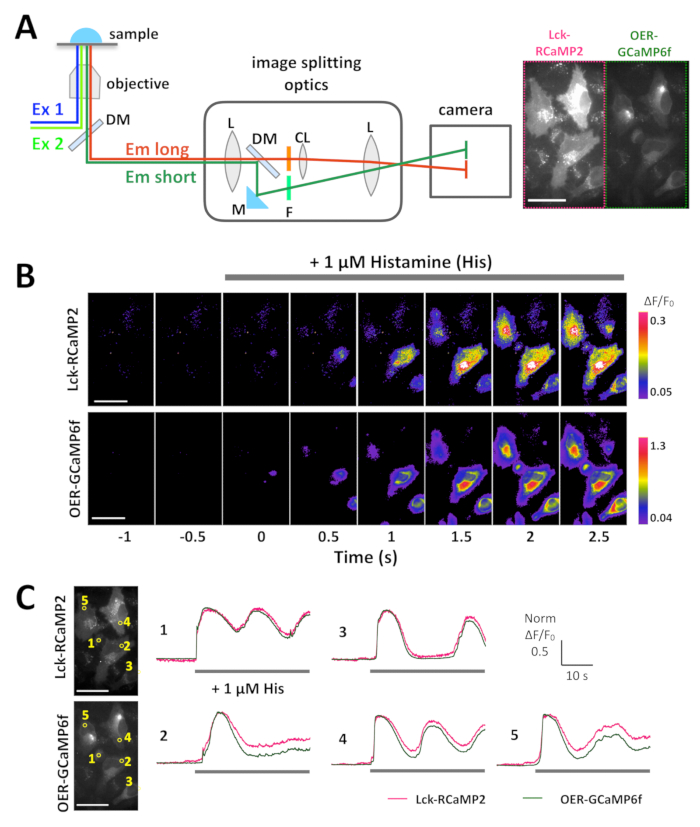
Figure 3: Example of the simultaneous imaging of Lck-RCaMP2 and OER-GCaMP6f in HeLa cells. (A) Schematic representation of signal separation with image-splitting optics. The same field of view for Lck-RCaMP2 and OER-GCaMP6f is simultaneously projected on the camera. A representative recording acquired at 10 Hz by a CMOS camera is provided in Video 1, and one frame of this recording is shown in panel A (right). (B) Pseudo-color images of ΔF/F0 for Lck-GCaMP2 (top) and OER-GCaMP6f (bottom). Histamine (His, 1 µM) was added at 0 s. (C) Representative normalized ΔF/F0 time course of Lck-GCaMP2 (magenta) and OER-GCaMP6f (green). Data were normalized to the maximum ΔF/F0 value for each plot. The gray bars indicate the timing of His application. Data were analyzed with a custom-made software TI Workbench15. Scale bar = 50 µm (microscopic image). Please click here to view a larger version of this figure.
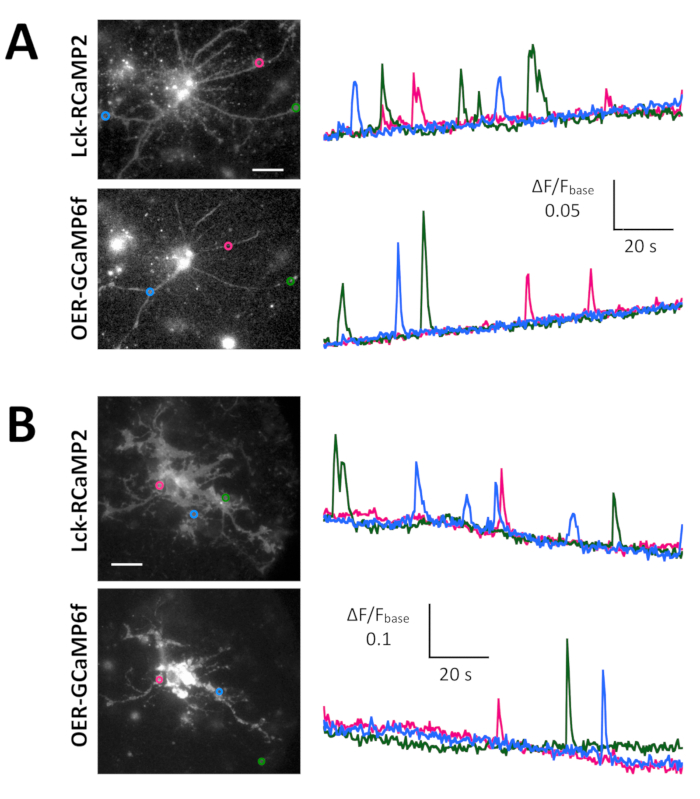
Figure 4: Spontaneous Ca2+ elevation in astrocytes monitored for Lck-RCaMP2 and OER-GCaMP6f expression. (A) Representative hippocampal and (B) cortical astrocytes transfected with Lck-RCaMP2 (top) and OER-GCaMP6f (bottom). Cortical cells were revived from frozen stock cultures. Lck-RCaMP2 and OER-GCaMP6f images were sequentially acquired in the same cell, at 2 Hz, with an EM-CCD camera. The plots on the left show the time courses of ΔF/Fbase measured in the ROIs indicated in the microscopic image. Data were analyzed with TI Workbench. Actual movies are provided in Video 2, Video 3, Video 4, and Video 5. The scale bar = 20 µm (microscopic image). The baseline drift suggests the changes in the global Ca2+ level in the cell. Please click here to view a larger version of this figure.
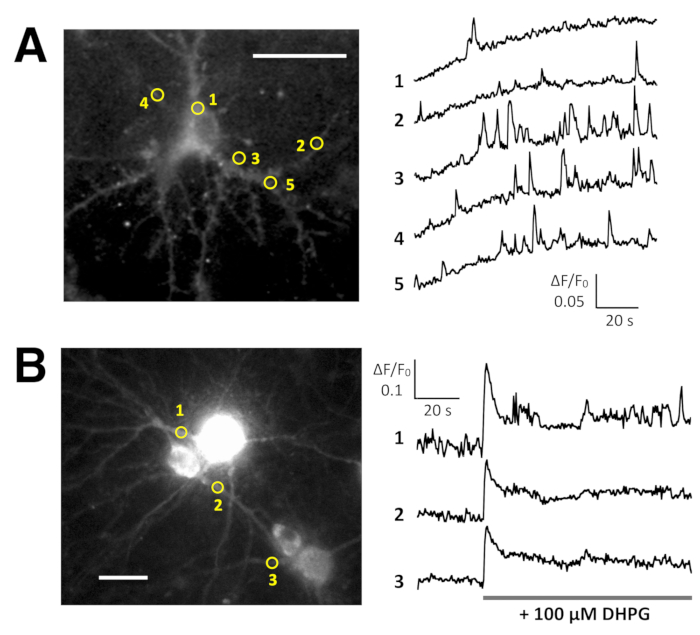
Figure 5: Examples of Ca2+ imaging in neurons with Lck-GCaMP6f and OER-GCaMP6f. (A) Representative rat hippocampal neurons expressing Lck-GCaMP6f at DIV 10 (left) and plots showing the time courses of ΔF/F0 measured in the ROIs (yellow circles) have been indicated in the image (right). The numbers in the time course correspond to the ROI numbers in the image. Note that the temporal pattern of Ca2+ elevation is different among the various regions of interest. The baseline drift suggests the increase in the global Ca2+ level in this neuron. (B) An example of mature mouse hippocampal neurons (DIV 30) infected with OER-GCaMP6f expression AAV vectors (left). The time course plot of ΔF/F0 measured shows the Ca2+ response to 100 µM (RS)-3,5-dihydroxyphenylglycine (DHPG) applied at the timing shown by the gray bar. Yellow circles show the position of ROIs where the time course was obtained. The images were acquired at 2 Hz with a cooled-CCD camera (panel A) or an EM-CCD camera (panel B) and analyzed with TI Workbench. Scale bar = 20 µm (microscopic image). Please click here to view a larger version of this figure.
Video 1: Example of simultaneous imaging of Lck-RCaMP2 and OER-GCaMP6f in HeLa cells. Representative recording acquired at 10 Hz and presented in Figure 3. Scale bar = 50 µm. Please click here to download this video.

Video 2: Spontaneous Ca2+ transient observed in Lck-RCaMP2 in hippocampal astrocyte. Representative recording acquired at 2 Hz (Figure 4A), recorded in the same field of view as Video 3. Scale bar = 20 µm. Please click here to download this video.
Video 3: Spontaneous Ca2+ transient observed in OER-GCaMP6f in a hippocampal astrocyte. Representative recording acquired at 2 Hz (Figure 4A), in the same field of view as Video 2. Scale bar = 20 µm. Please click here to download this video.
Video 4: Spontaneous Ca2+ transient observed in Lck-RCaMP2 in a cortical astrocyte Representative recording acquired at 2 Hz (Figure 4B), recorded in the same field of view as Video 5. Scale bar = 20 µm. Please click here to download this video.
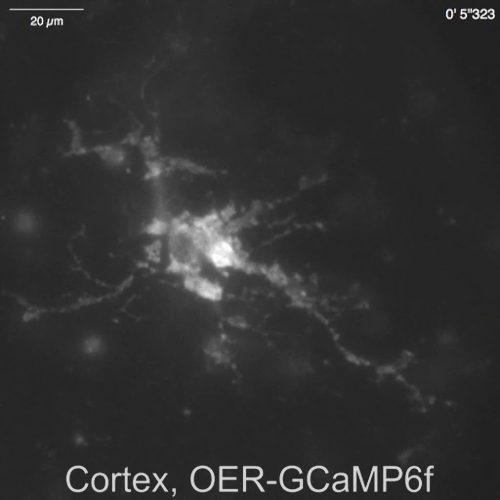
Video 5: Spontaneous Ca2+ transient observed in OER-GCaMP6f in a cortical astrocyte. Representative recording acquired at 2 Hz (Figure 4B), in the same field of view as Video 4. The scale bar = 20 µm. Please click here to download this video.
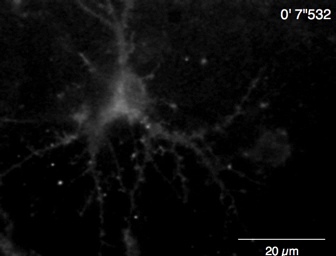
Video 6: Example of Ca2+ imaging in a rat hippocampal neuron (DIV 10) by Lck-GCaMP6f. Example of neuronal Ca2+ signals recorded at 2 Hz (Figure 5A). Scale bar = 20 µm. Please click here to download this video.
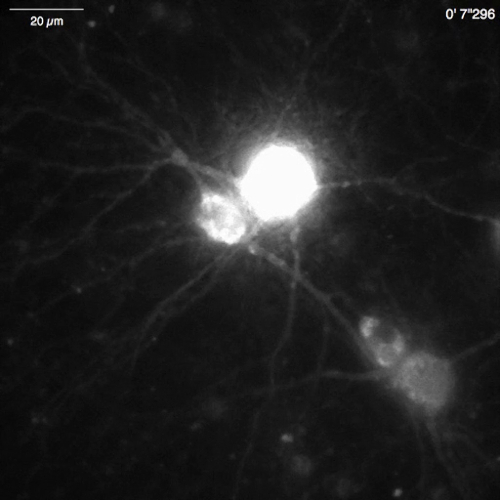
Video 7: Ca2+ release in a mouse hippocampal neuron (DIV 30) expressing OER-GCaMP6f. Example of neuronal Ca2+ signals recorded in a mouse hippocampal neuron infected with OER-GCaMP6f expression AAV vectors (Figure 5B). The neuron was stimulated with 100 µM dihydroxyphenylglycine (DHPG) applied at 30 s to evoke Ca2+ release from the ER. Scale bar = 20 µm. Please click here to download this video.
