The protocol described in this manuscript allowed isolation of a nearly homogenous population of HDFs (Figure 1). As demonstrated by immunofluorescent staining, almost 100% of the human dermal cells isolated using the conditions described in this protocol were positively stained for dermal fibroblast markers, vimentin, and collagen I24, but negative for human foreskin keratinocyte marker K14 (Figure 1). Figure 2 shows the process of generating MCPyV virions using recombinant MCPyV genome and a virion sample after ultracentrifuge concentration. After visualizing the band of MCPyV virions concentrated in the core of the gradient (marked by an arrow in Figure 2B), 500 µL fractions were collected and MCPyV qPCR was performed to identify the peak fractions. An example of the qPCR analysis data is shown in Figure 2C. In some other experiments, the samples were also analyzed with Western blotting using a rabbit-anti-MCPyV VLP antibody (Supplemental Figure 1). Figure 3 shows immunofluorescent stained images of HDFs infected with MCPyV. To manually quantify positive cells, we counted at least three random views of about 300 cells. We consider cells that are LT positive, VP1 positive, or both LT and VP1 positive to be positive for MCPyV infection. In the experiments shown in Figure 3, over 30% of cells are LT positive and more than 10% are VP1 positive. The MCPyV genomes replicated in the infected cells were detected using both the HCR-DNA FISH (Figure 4A) and Immunofluorescent-HCR-DNA FISH (Figure 4B). While the HCR-DNA FISH reveals the localization of MCPyV DNA present in the replication factory (foci) in Figure 4A, the Immunofluorescent-HCR-DNA FISH methods allows simultaneous detection of both MCPyV DNA and LT protein co-localizing at the replication centers (Figure 4B). The images from the immunofluorescent-HCR-DNA FISH demonstrated that this technique could be used to reveal co-localization of viral DNA and the associated viral protein.
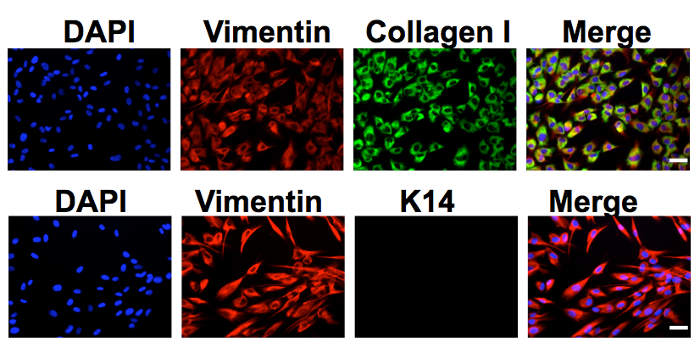
Figure 1. Human dermal fibroblasts isolated from neonatal human foreskin. Human dermal cells cultured in DMEM/F12 medium supplemented with 10% FBS were stained using antibodies against vimentin, collagen I, or keratin 14 (K14). The cells were also counterstained with DAPI. Bar, 50 µm. This figure was adapted from Figure S2 of Liu et al., 201624. Please click here to view a larger version of this figure.
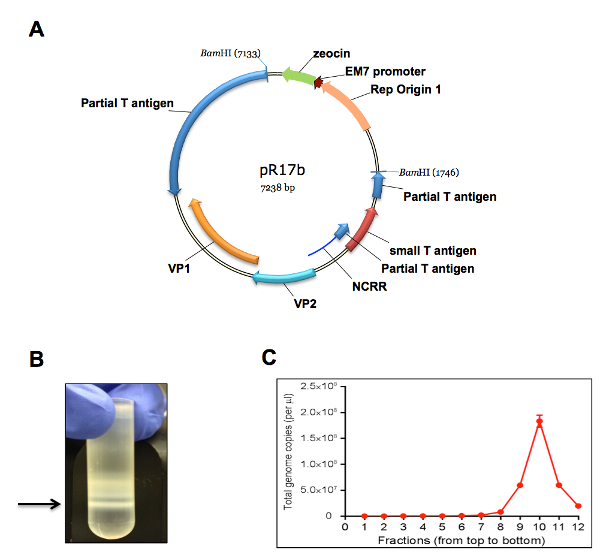
Figure 2. Production of MCPyV virion using recombinant viral genome. (A) A plasmid map of pR17b (MCPyV genome plasmid). (B) A representative picture of an MCPyV virion sample harvested and purified over a gradient. Arrow marks the band of MCPyV virions concentrated in the core of the gradient. (C) The viral genome copy number in each gradient fraction was quantified using qPCR. Core gradient fractions (numbers, counting from the top of the gradient, are indicated at the bottom of the graph). Error bars represent standard error of the mean (S.E.M.) of three technical repeats. Please click here to view a larger version of this figure.
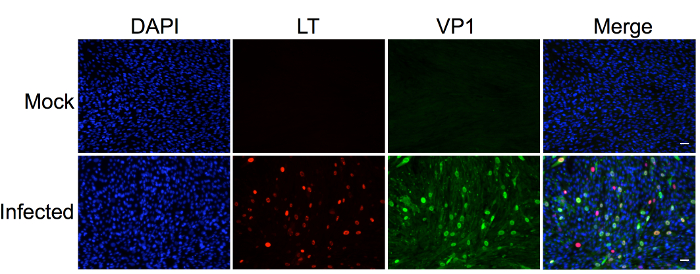
Figure 3. Human dermal fibroblasts support robust MCPyV infection, transcription, and replication. Dermal fibroblasts isolated from human skin were treated with MCPyV virions in DMEM F12 medium containing EGF, bFGF, CHIR99021, and collagenase IV for two days. After changing to fresh DMEM/F12 medium containing 20% FBS for three more days, cells were immuno-stained using the indicated antibodies and counterstained with DAPI. Many of cells not only have highly expressed LT and VP1 but also show robust MCPyV replication foci. This figure was adapted from Figure 3 of Liu et al., 201624. Scale bar, 50 m. Please click here to view a larger version of this figure.
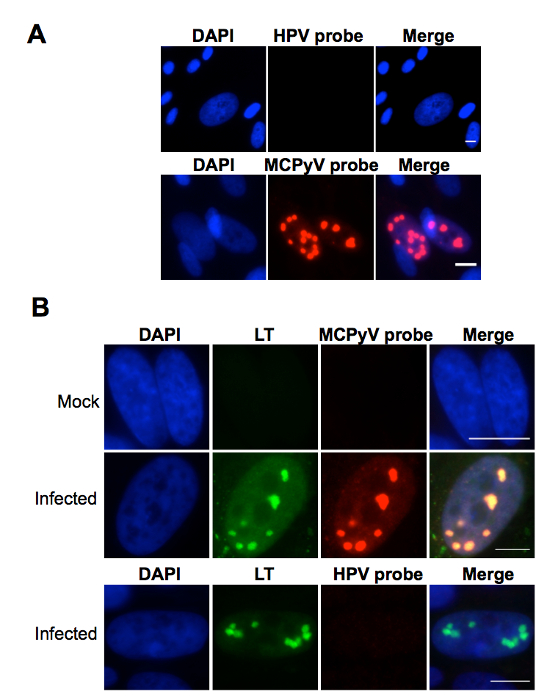
Figure 4. Detection of MCPyV in infected cells using FISH techniques. (A) Human dermal cells cultured in DMEM/F12 containing EGF, bFGF, CHIR99021, and collagenase type IV were treated with MCPyV for 2 days. After changing to fresh DMEM/F12 containing FBS for 3 more days, the cells were subjected to HCR-DNA FISH analysis. The cells were also counterstained with DAPI. (B) Human dermal cells cultured in medium containing EGF, bFGF, and CHIR99021 were either untreated (Mock) or treated (Infected) with MCPyV as described in (A). The cells were subjected to immuno-FISH using LT antibody and MCPyV-specific DNA probes before counterstaining with DAPI. HPV16 probes were used as a negative control. Scale bar, 10 m. This figure was adapted from Figure 4 of Liu et al., 201624. Please click here to view a larger version of this figure.
| Probe name | Sequences |
| MCPyV probe 1 | CCTCAACCTACCTCCAACTCTCACCATATTCGCTTC TAAAA tgagctacctcactaaggagtggtttttatactgcagtttcccgcccttg ATTTT CACATTTACAGACCTCAACCTACCTCCAACTCTCAC |
| MCPyV probe 2 | CCTCAACCTACCTCCAACTCTCACCATATTCGCTTC TAAAA agaggcctcggaggctaggagccccaagcctctgccaacttgaaaaaaaa ATTTT CACATTTACAGACCTCAACCTACCTCCAACTCTCAC |
| MCPyV probe 3 | CCTCAACCTACCTCCAACTCTCACCATATTCGCTTC TAAAA cattgactcatttcctggagaggcggagtttgactgataaacaaaacttt ATTTT CACATTTACAGACCTCAACCTACCTCCAACTCTCAC |
| MCPyV probe 4 | CCTCAACCTACCTCCAACTCTCACCATATTCGCTTC TAAAA gatactgccttttttgctaattaagcctcttaagcctcagagtcctctct ATTTT CACATTTACAGACCTCAACCTACCTCCAACTCTCAC |
| MCPyV probe 5 | CCTCAACCTACCTCCAACTCTCACCATATTCGCTTC TAAAA aagcttctcctgtaagaatagcttccaaagttactcctgtggtggcactt ATTTT CACATTTACAGACCTCAACCTACCTCCAACTCTCAC |
| MCPyV probe 6 | CCTCAACCTACCTCCAACTCTCACCATATTCGCTTC TAAAA ggatgttgccataacaattaggagcaatctccaaaagcttgcacagagcc ATTTT CACATTTACAGACCTCAACCTACCTCCAACTCTCAC |
| MCPyV probe 7 | CCTCAACCTACCTCCAACTCTCACCATATTCGCTTC TAAAA gctcaggggaggaaagtgattcatcgcagaagagatcctcccaggtgcca ATTTT CACATTTACAGACCTCAACCTACCTCCAACTCTCAC |
| MCPyV probe 8 | CCTCAACCTACCTCCAACTCTCACCATATTCGCTTC TAAAA aagcctgggacgctgagaaggacccatacccagaggaagagctctggctg ATTTT CACATTTACAGACCTCAACCTACCTCCAACTCTCAC |
| MCPyV probe 9 | CCTCAACCTACCTCCAACTCTCACCATATTCGCTTC TAAAA agcttcgggaccccccaaattttcgctttcttgagaatggaggaggggtc ATTTT CACATTTACAGACCTCAACCTACCTCCAACTCTCAC |
| MCPyV probe 10 | CCTCAACCTACCTCCAACTCTCACCATATTCGCTTC TAAAA cttttggctagaacagtgtctgcggcttgttggcaaatggttttctgaga ATTTT CACATTTACAGACCTCAACCTACCTCCAACTCTCAC |
| HPV probe 1 | CCTCAACCTACCTCCAACTCTCACCATATTCGCTTC TAAAA cagctctgtgcataactgtggtaactttctgggtcgctcctgtgggtcct ATTTT CACATTTACAGACCTCAACCTACCTCCAACTCTCAC |
| HPV probe 2 | CCTCAACCTACCTCCAACTCTCACCATATTCGCTTC TAAAA acaatattgtaatgggctctgtccggttctgcttgtccagctggaccatc ATTTT CACATTTACAGACCTCAACCTACCTCCAACTCTCAC |
| HPV probe 3 | CCTCAACCTACCTCCAACTCTCACCATATTCGCTTC TAAAA gtcagctatactgggtgtaagtccaaatgcagcaatacaccaatcgcaac ATTTT CACATTTACAGACCTCAACCTACCTCCAACTCTCAC |
| HPV probe 4 | CCTCAACCTACCTCCAACTCTCACCATATTCGCTTC TAAAA ctttggtatgggtcgcggcggggtggttggccaagtgctgcctaataatt ATTTT CACATTTACAGACCTCAACCTACCTCCAACTCTCAC |
| HPV probe 5 | CCTCAACCTACCTCCAACTCTCACCATATTCGCTTC TAAAA ccatccattacatcccgtaccctcttccccattggtacctgcaggatcag ATTTT CACATTTACAGACCTCAACCTACCTCCAACTCTCAC |
Table 1: Probes used in the study.
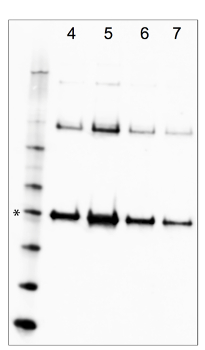
Supplemental Figure 1. Screening Iodixanol gradient fractions for MCPyV VP1. MCPyV-infected cells were lysed and subjected to Iodixanol gradient fractionation. Core gradient fractions (numbers, counting from the bottom of the gradient in this experiment, are indicated at the top of the figure) were subjected to Western blot analysis to detect VP1. 10 µL samples of each fraction were adjusted to 1x load dye with a 5% final concentration of 2-mercaptoethanol and briefly heated to 65 °C. The samples were separated on a 4-12% bis-Tris gel and blotted onto nitrocellulose. Western blotting was conducted in TBST with nonfat dry milk using a rabbit-anti-MCPyV VLP antiserum diluted 1:5000. The antiserum is available upon request. The 50 kDa standard (which migrates similarly to the 47 kDa MCPyV VP1 monomer) is marked with an asterisk. In the shown image, sample reduction was incomplete and disulfide-linked VP1 multimers are apparent. Use of dithiothreitol and/or higher denaturation temperatures can result in more complete reduction to the VP1 monomer species. Please click here to view a larger version of this figure.
