All mice were housed in a humidity- and temperature-controlled, AAALAC-accredited animal facility at the University of Miami Miller School of Medicine. All experiments were approved by the University of Miami Miller School of Medicine Institutional Animal Care and Use Committee (IACUC) and conducted according to specifications of the NIH.
1. Preparation of the Sample Extract
- Adherent cells
- Plate cells in 10 cm dishes in the appropriate culture media (1 x 106 to 1 x 109 cells per dish for cell lines, such as BV2, HEK-293, and SH-SY5Y, but ~1 x 1015 cells per dish for primary cells, such as primary cortical neurons). Assure that the cells are equally distributed on the entire surface of the plate and allow the cells to grow for 48 h to reach ~100% confluence (37 °C, 5% CO2).
- Once the cells have reached the desired confluency, gently aspirate the culture media and wash the cells 2x with prewarmed serum-free media under a tissue culture hood.
- Aspirate the serum-free media from the dish and add 1 mL of ice-cold extraction buffer (0.4 M sulfuric acid, 1 mM KCl, 1 mM MgCl2, 50 mM Tris-HCl [pH 8.0] and 1x protease inhibitor cocktail) to each dish.
- Use a plastic cell scraper to collect all cells in the extraction buffer (by scraping) and transfer them to a 1.5 mL labeled tube with a 1,000 µL pipette. Pipet the cells up and down 3x to facilitate homogenization.
- Close all tubes and immediately put them on ice.
- Brain tissue
- If frozen tissue is being used, place the tissue in a prechilled 1.5 mL tube and briefly thaw it on ice. If fresh tissue is being used, proceed immediately to step 1.2.2.
NOTE: The current protocol describes procedures using frozen mouse brain and mouse prefrontal cortex samples. - Homogenize the tissue using a handheld Dounce homogenizer using the appropriate amount of extraction buffer and the recommended number of strokes (Table 1). To avoid excessive chromatin shredding, do not exceed the number of recommended strokes.
- Using a single channel 1,000 µL pipette, transfer the homogenate to a prechilled 1.5 mL tube. Close all tubes and immediately put them on ice.
- If frozen tissue is being used, place the tissue in a prechilled 1.5 mL tube and briefly thaw it on ice. If fresh tissue is being used, proceed immediately to step 1.2.2.
2. Preparation of the Crude Histone Extract
- Place the 1.5 mL tubes containing the cells or tissue suspended in extraction buffer on a rotating platform and rotate at 15 rpm at 4 °C to allow the extraction of crude histones.
NOTE: The extraction time may vary for different cell and tissue types and must be optimized for each procedure. The current protocol presents results obtained following 15 min, 2 h, and 24 h of extraction (Figure 2, Figure 3, Figure 4, and Figure 5). - Prechill the microcentrifuge to 4 °C. After the desired extraction time has passed, centrifuge the tubes at maximum speed for 10 min at 4 °C.
- Transfer the supernatant including the crude histones to a new, prechilled 1.5 mL tube. Discard the pellet.
- Store the supernatant at -80 °C (the extraction can be stopped at this step, see “Stop step” in Figure 1) or immediately proceed to the next step.
- Neutralize the crude histones with a 1/4 volume of 5x neutralization buffer (e.g., add 250 µL of 5x neutralization buffer to 1 mL of crude histones). Mix well by pipetting up and down 6x.
- Check the pH of the mixture with pH strips. Adjust accordingly by adding more neutralization buffer to reach a pH of 7.
- Evaluate the presence of histone and nonhistone protein in the crude histone extract as follows (Figure 2).
- Add 37.5 µL of the sample to 12.5 µL of 4x (Laemmli) sample buffer and denature for 10 min at 99 °C.
- Load the sample onto an SDS-PAGE gel and run the gel for 1 h at 100 V.
- Stain the gel overnight with Coomassie Brilliant Blue R-250 staining solution and destain during three consecutive washes (1 h/wash) with Coomassie Brilliant Blue R-250 destaining solution.
NOTE: Crude histones (Figure 2) can be compared with eluted and purified histones (i.e., column input [Figure 5A]).
3. Purification of Core Histones
- Spin column equilibration
- Add 500 µL of equilibration buffer to each spin column being used. Do not touch the column membrane.
- Centrifuge at 4 °C for 3 min at 800 x g. Discard the flow-through. Repeat 1x.
- Histone Purification
- Add 500 µL of the sample of interest from step 2.6 to the column. Centrifuge at 4 °C for 3 min at 800 x g. Collect the flow-through.
- Repeat the previous step as many times as necessary to load the entire sample onto the column. Do not overfill the spin column.
- Combine the flow-through from each centrifugation step to analyze the column-binding efficiency (Figure 3).
- Follow step 2.7 to analyze the column flow-through.
- Column wash
- Add 500 µL of the wash buffer to each column. Centrifuge at 4 °C for 3 min at 800 x g. Collect the flow-through wash (wash #1).
- Repeat step 3.3.1 for a total of three washes. Collect the flow-through washes #2 and #3. Do not pool the consecutive column flow-through washes.
- To further evaluate the column’s histone-binding efficiency, analyze the three column washes by following step 2.7 (Figure 4).
- Histone elution
- Transfer the column into a new labelled 1.5 mL tube.
- Add 50 µL of the histone elution buffer. Centrifuge at 4 °C for 3 min at 800 x g. Save the flow-through containing histone proteins.
- For an additional elution, repeat step 3.4.2. Do not combine the first and second flow-through eluates as they differ in histone quantity and purity.
4. Precipitation of Core Histones
- Add perchloric acid (PCA) to the purified histones to a final concentration of 4% PCA (e.g., add 3 µL of 70% PCA to 50 µL of purified histones from step 3.4.2.
- Centrifuge for 3 s to collect all residual liquid from the tube wall. Mix by pipetting up and down 6x.
- Place the tubes in a rack and incubate for 24 h at 4 °C.
- The next day, prechill a microcentrifuge to 4 °C and centrifuge the samples for 75 min at maximum speed at 4 °C.
- After the centrifugation is complete, a small white pellet containing precipitated histones will be visible on the bottom of the tube. Do not vortex the sample.
- Carefully aspirate the supernatant and, without disturbing the pellet, add 500 µL of ice-cold 4% PCA to the sample.
- Centrifuge at 4 °C for 10 min at maximum speed. Carefully aspirate the supernatant.
- Repeat step 4.7 2x.
- Without disturbing the pellet, add 500 µL of ice-cold acetone. Centrifuge at 4 °C for 10 min at maximum speed. Carefully aspirate the supernatant.
- Repeat step 4.9 2x.
- Carefully aspirate the supernatant, leave the tubes uncapped, and allow the sample to dry on ice for 30 min. Check if all residual acetone has evaporated.
- Leave the tubes uncapped and allow the sample to dry at room temperature (RT) for 5 min.
- Resuspend the pellet in 30 µL of sterile water. Do not pipet up and down. Flick the tube gently with a finger.
- Cap all tubes and allow the histones to reconstitute on ice for 30 – 50 min, depending on the pellet size. Check if the pellet is resuspended.
- Cap all tubes and allow the pellet to further resuspend at RT for 5 min.
NOTE: This solution (first and second elution from step 3.4.3) is constituted of purified and desalted histones and may be used for further quantification and histone acetylation analysis.
5. Quantification of Eluted Histone Proteins
- Use a spectrophotometer according to the manufacturer’s protocol to quantify the total histone proteins obtained after the final elution in step 4.15. Measure the absorbance at 230 nm. Record the A260/A280 ratio indicative of the sample contamination with nucleic acid.
- Use the following formula to calculate the histone concentration (x):
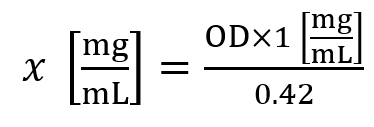
Here, OD is the optical density measured at A230 nm. - A histone concentration of ~1.5 mg/mL is considered an average yield for cell lines, while a histone concentration of ~5.0 mg/mL is considered an average yield for 30 mg of tissue.
6. Western Blot Analysis
- Adjust the purified and eluted histone from step 4.15 to ~10 µg of histone protein/sample.
- Add the appropriate volume of water and 4x Laemmli sample buffer to adjust the loading volumes.
- Denature the samples for 10 min at 99 °C. Cool them on ice. Centrifuge for 3 s to collect all residual liquid and condensation from the tube wall.
- Load the samples onto an SDS-PAGE gel and run the gel for 1 h at 100 V.
- To visualize total histone protein, stain the gel overnight with Coomassie Brilliant Blue R-250 staining solution and destain during three consecutive washes (1 h/wash) with Coomassie Brilliant Blue R-250 destaining solution.
NOTE: The first elution of histone proteins contains high-quality histones (Figure 5A) while the second elution contains low to no levels of histones (Figure 5B). - To quantify histone PTMs, use a transfer system (see Table of Materials) to transfer the histone protein from the SDS-PAGE gel (step 6.4) onto a PVDF membrane.
- To assemble the transfer sandwich, open the transfer system cassette and place the PVDF membrane stack (labelled as Bottom+) on the bottom of the cassette with the membrane facing up. Roll the membrane gently with a blot roller to remove any air from between the stack and the membrane.
- Lay the gel on top of the membrane, roll the gel gently with a blot roller to remove any air from between the membrane and the gel, and place the top stack on the gel. Roll gently again and place the cassette cover on top of the sandwich, press down firmly, and turn the nob clockwise to lock.
- Insert the cassette to the transfer system slot. On the apparatus screen, select Turbo Protocol. Use a 3 min protocol for a single mini gel or a 7 min protocol for more than two mini gels.
- Stain the membrane with Ponceau S stain for 5 min and visualize the total histone proteins.
- Wash them in 1x Tris-buffered saline (TBS) with 0.1% Tween 20 for 2 h and block them in 5% milk for 1 h. Incubate with primary and secondary antibody (overnight at 4 °C or 1 h at RT, respectively) or according to a previously optimized protocol.
NOTE: In the current protocol, antibodies against acetylated histones H4K12 (Figure 6 and Figure 7) and H3K27 (Figure 8) were used.
To illustrate the progression of the histone purification protocol and composition of all analyzed fractions, we evaluated different histone extracts from human microglial BV2 cells. To demonstrate the quantification of histones H3 and H4 PTMs (i.e., acetylation), we used brain tissue lysates.
BV2 cells were plated at 5 x 106 cells per dish in 10 cm tissue culture-treated dishes and allowed to grow to confluency for 48 h. Cells were then collected, and histones were released from chromatin by an incubation in extraction buffer containing 0.4 M sulfuric acid, 1 mM KCl, 1 mM MgCl2, 50 mM Tris-HCl (pH 8.0), and 1x protease inhibitor. The extraction time, between 15 min and 24 h, did not affect the overall composition of crude histone extracts as determined by Coomassie Brilliant Blue staining (Figure 2). Next, crude histones were passed through the equilibrated histone columns and the flow-through was analyzed. High column-binding efficiency is determined by the absence of histone protein in the flow-through when analyzed by Coomassie Brilliant Blue staining. We determined the column-binding efficiency to be 100% as there were no detectable histone proteins present in the analyzed flow-through (Figure 3). All membranes with bound histones were then washed three times with wash buffer to remove any remaining impurities, leaving only histone proteins bound to the silica gel. We determined that, for all histone extraction times (i.e.,15 min, 2 h, and 24 h), the first membrane wash was the most important to remove nonhistone contaminations from the columns, while the second and third washes did not influence the sample purity. Thus, depending on the sample type, the last two washes might be omitted. Following the first elution of histone protein from the column (using elution buffer containing 1mM NaCl and EDTA), histones were precipitated overnight with 4% perchloric acid and then pelleted, washed, and analyzed for the enrichment of purified histones H3 and H4. We observed that 24 h of extraction time increases the amount of H3 and H4 histones in the purified fraction compared to 15 min and 2 h of extraction time (Figure 5A). The second elution from the column did not result in high-quality or high-quantity histones (Figure 5B).
Next, we used brain tissue homogenates to quantify histone H3 and H4 PTMs, namely acetylation. Wild-type (C57BL6/J) male mice were administered a broadly acting HDAC inhibitor (tributyrin) at a dose of 5 g/kg orally for 3 d. Whole-brain tissue was collected on day 4 and crude histones were extracted according to the described protocol. Using an unpaired t-test, we determined that tributyrin increases the acetylation of histones in the crude extract (t(6) = 6.184, P = 0.0004); however, impurities are detected in the extract (the histone bands are not clearly defined). Thus, H4K12ac antibody does not have a high specificity (Figure 6). To further evaluate the applicability of the presented protocol to smaller tissue sections, we collected the prefrontal cortex from triple transgenic Alzheimer's disease (3 x Tg-AD) mice treated daily with 10 mg/kg M344, a class I and IIb HDAC inhibitor, for four months. Histone purification and precipitation was performed according to the herein described protocol. Using the purified histone H3 and H4 fraction, we determined that M344 increases H4K12 acetylation 2.4-fold (t(6) = 13.03, P < 0.0001), with a high specificity of H4K12ac antibody (Figure 7). Similarly, we observed an increase in histone H3 acetylation in BV2 cells in response to another HDAC inhibitor, namely the selective HDAC3 inhibitor, RGFP-966. 10 µM of RGFP-966 causes an approximately twofold increase of acetylation at histone H3K27 after 24 h of treatment. Student's unpaired t-test was used to compare control versus treated cells.
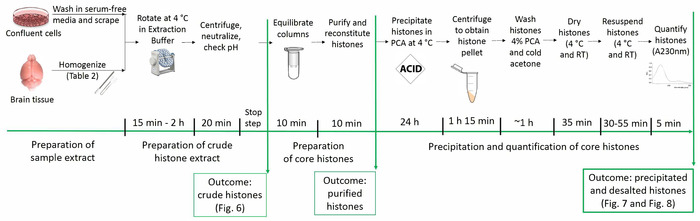
Figure 1: Timeline of the histone purification protocol. All steps for histone analyses are shown below along with the estimated time needed for each step. Figures depicting the outcome of particular steps and presented within the manuscript are referred to in parentheses. Please click here to view a larger version of this figure.
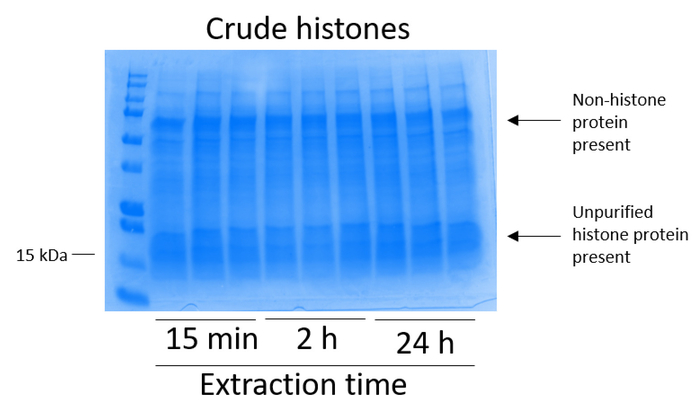
Figure 2: Representative Coomassie Brilliant Blue-stained gel demonstrating crude histones extracted from BV2 cells. BV2 cells were cultured for 48 h before the histone extraction protocol began. Crude histones were extracted for 15 min, 2 h, and 24 h, with three replicates for each time point (this is also the case in Figure 3, Figure 4, and Figure 5). Both nonhistone and histone proteins are present in the crude histone extract. Please click here to view a larger version of this figure.
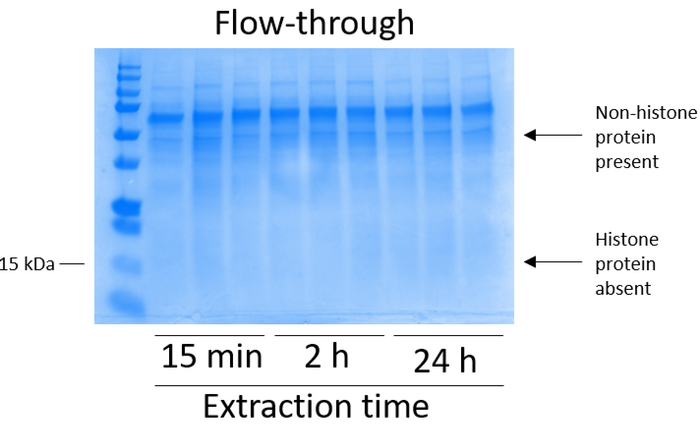
Figure 3: Representative Coomassie Brilliant Blue-stained gel demonstrating column flow-through following histone purification steps from BV2 cells. Following crude histone passing through the histone binding column, only nonhistone proteins are present in the flow-through. Histone proteins are absent in this fraction. Please click here to view a larger version of this figure.
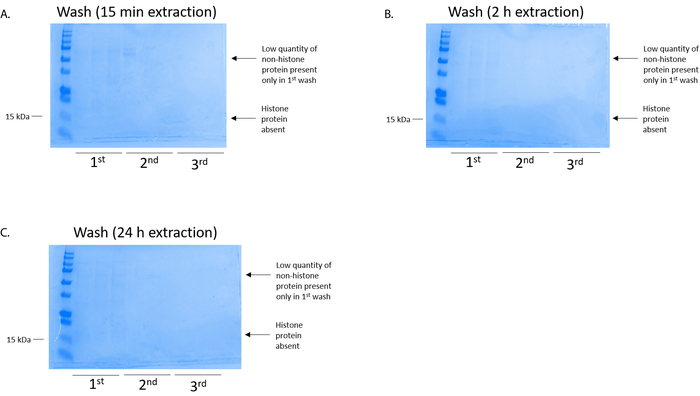
Figure 4: Representative Coomassie Brilliant Blue-stained gel demonstrating a column wash following a histone purification step from BV2 cells. Regardless of the histone extraction times, which were (A) 15 min, (B) 2 h, or (C) 24 h, low quantities of nonhistone proteins were only present in the first-wash histonebinding column. Histone proteins were absent in all washes. Please click here to view a larger version of this figure.
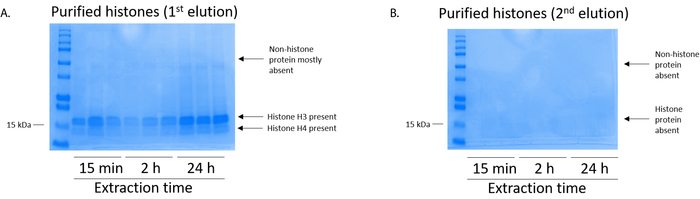
Figure 5: Representative Coomassie Brilliant Blue-stained gel demonstrating elutions following a histone purification step from BV2 cells. (A) High-quality purified and desalted histone H3 and H4 were detected after the first elution from the histone purification column. (B) The second elution from the histone purification column did not yield a high quality or quantity of histones H3 or H4. Please click here to view a larger version of this figure.
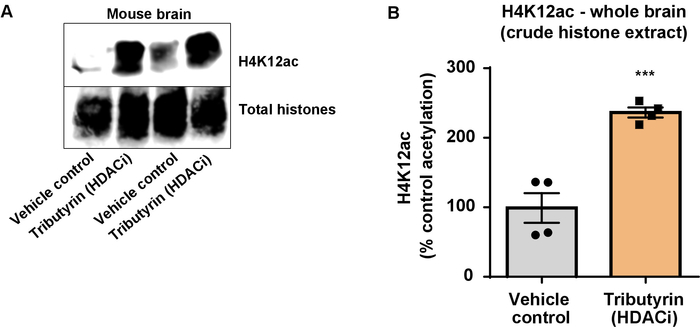
Figure 6: The broadly acting HDAC inhibitor, tributyrin, increases H4K12 acetylation in the whole brain of wild-type mice. (A) This panel shows a representative western blot depicting an increase of H4K12 acetylation in the crude histone extract collected from the whole brain of wild-type mice in response to the broadly acting HDAC inhibitor, tributyrin. (B) This panel shows the quantification of the increase in H4K12 acetylation in vivo. Unpaired t-test was used to compare groups (t(6) = 6.076, P = 0.0005). The bars represent the mean ± the standard error of the mean (SEM). N = 8. ***P < 0.0001. Please click here to view a larger version of this figure.
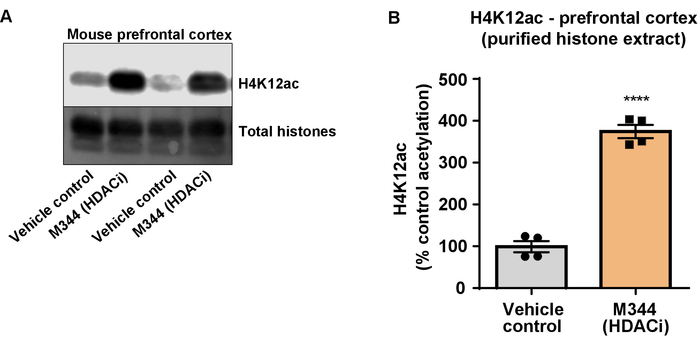
Figure 7: The class I and IIb HDAC inhibitor, M344, increases H4K12 acetylation in the prefrontal cortex of triple transgenic Alzheimer's disease (3 x Tg-AD) mice. (A) This panel shows a representative western blot depicting an increase of H4K12 acetylation in the purified and desalted histone extract collected from the prefrontal cortex of 3 x Tg-AD mice in response to the inhibition of HDACs by M344. (B) This panel shows the quantification of the increase of H4K12 acetylation in response to M344 administered at a 10 mg/kg daily dose for four months. Unpaired t-test was used to compare groups (t(6) = 13.30, P < 0.0001). The bars represent the mean ± the SEM. N = 8. ****P < 0.00001. Please click here to view a larger version of this figure.
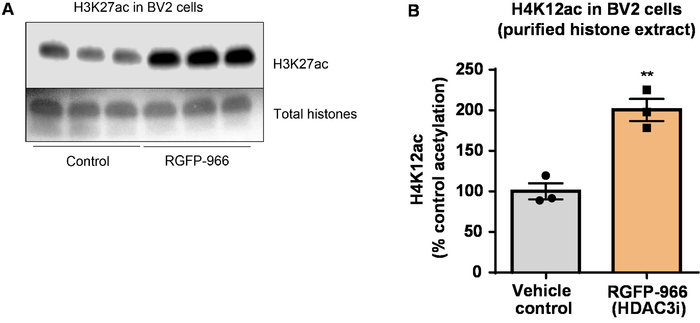
Figure 8: The selective HDAC3 inhibitor, RGFP-966, increases H3K27 acetylation in BV2 microglial cells. (A) This panel shows a representative western blot depicting an increase of H3K27 acetylation in the purified and desalted histone extract collected from BV2 cells in response to the HDAC3 inhibition by RGFP-966. (B) RGFP-966 causes an approximately twofold increase of acetylation at histone H3 and lysine (K) 27 after 24 h of treatment. Unpaired t-test was used to compare control versus treated cells (t(4) = 5.981, P = 0.002). The bars represent the mean ± the SEM. N = 6. **P < 0.01. Please click here to view a larger version of this figure.
| Representative tissue (one hemisphere) * | Average tissue weight (mg) | Extraction Buffer (mL) | Number of strokes |
| Mouse cerebellum | 40 | 1 | 40 |
| Mouse frontal cortex | 30 | 0.3 | 20 |
| Mouse hippocampus | 27 | 0.3 | 18 |
| Mouse entorhinal cortex | 19 | 0.3 | 17 |
| * All experiments were performed on adult male mice. | |||
| Average age: 16 months. Average weight: 30 grams. | |||
Table 1: Optimized condition for brain tissue homogenization.
