As shown in Figure 4, this procedure allows excellent imaging of GFP-labeled axons in adult Drosophila legs, together with their terminal arbors. Importantly a clean GFP signal is obtained without any contamination from the fluorescence emitted by the leg cuticle. The signal from the cuticle can then be combined with the GFP signal to identify the positioning of axons in the legs (Figure 4E, Figure 1,and Video 1). Critically, it is important to obtain well-fixed legs. Figure 5 shows examples of a well-fixed (Figure 5A) and a badly-fixed leg (Figure 5B). In the former case the internal structures inside the legs are of a uniform color and the tracheas, which are dark, are visible. A main tracheal tube runs in the center of each leg segment (adjacent to the main nerve trunk) and many thinner ramifications are also visible. In the latter, dark material is present in the tarsus and tibia and the tracheal system is not clearly visible in the femur and coxa: in such cases it is always observed that the signal from fluorescent proteins is degraded being of low intensity or absent altogether. Second, careful dissection of the legs is necessary to obtain all the leg segments (from coxa to tibia) and to avoid mechanical shock to the legs. Third, the legs must be left in mounting medium long enough for it to penetrate the inside of the legs. Sometimes leg segments, especially the femur, appear collapsed — this can be due to lack of penetration of the fixative and/or mounting medium. Finally, one must use high quality microscope objectives, corrected for flatness of field and specially designed for fluorescence and/or apochromatic.
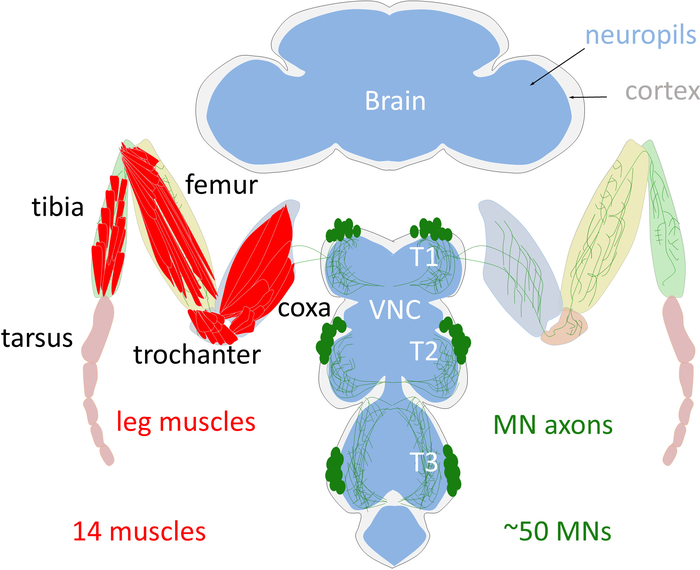
Figure 1: Schema of the adult Drosophila leg motor system. The cell bodies of the adult leg MNs (green) are localized in the cortex (grey) of the thoracic ganglion of the VNC. MNs arborize their dendrites in the leg neuropil (blue) and send their axons into the leg to innervate one of the 14 leg muscles (red). Note that only the T1 legs are schematized. Please click here to view a larger version of this figure.
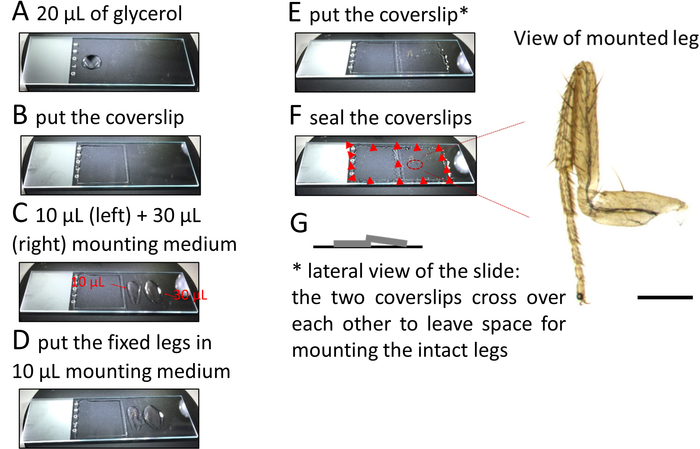
Figure 2: Procedure to mount legs on microscope slides. Please click here to view a larger version of this figure.
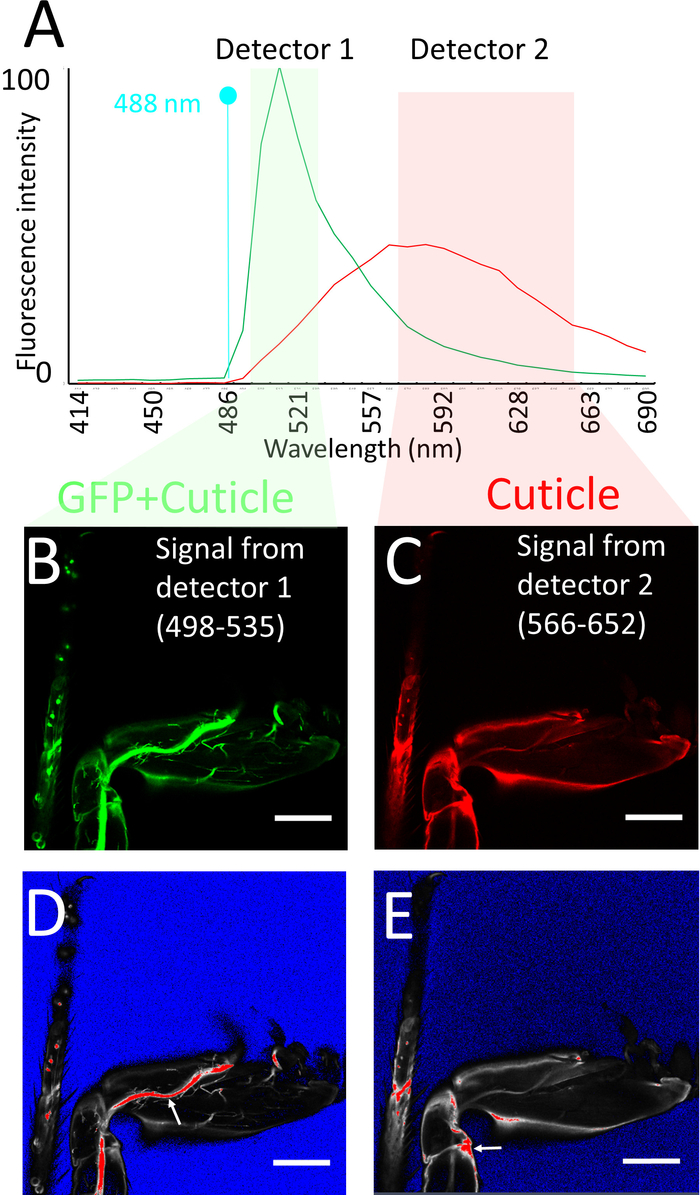
Figure 3: Imaging procedure. (A) Emission spectra of GFP and of leg cuticle using 488 nm Argon laser excitation, obtained using spectral imaging of sections of legs expressing GFP and of legs of Drosophila that do not express GFP respectively. Note that the fluorescence intensities have not been normalized, e.g., are from raw data using the same parameters (objective, mounting medium, laser power, confocal aperture, gain, offset) as for the leg imaging procedure. Also shown are the detector windows used for imaging the GFP + cuticle fluorescence (detector 1: green) and cuticle (detector 2: pink), based on the GFP vs cuticle background spectra. (B, C) Confocal section of legs labeled with mCD8::GFP under the control of DVGlut-Gal4 obtained from detector 1 (B) and detector 2 (C). (D, E) Saturation marker settings used to image (B, C) respectively (see text for explanation): blue no signal, red saturation. Note that on detector 1 the main nerve tracks are saturated, while on detector 2 some regions of the cuticle are saturated (see arrows). Scale bar = 100 µm. Please click here to view a larger version of this figure.
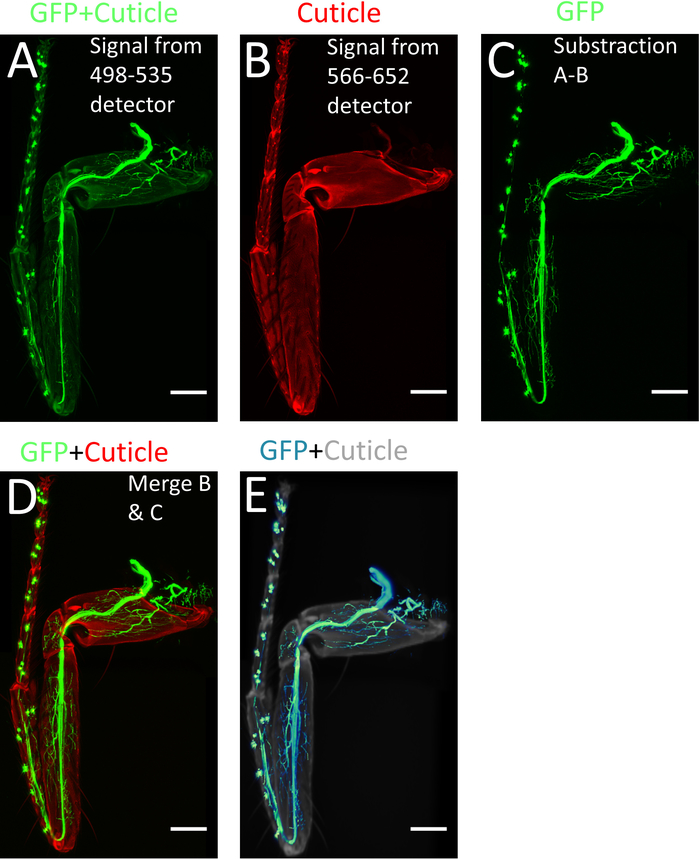
Figure 4: Processing of images using ImageJ/FIJI. (A) Max projection of the confocal stacks obtained from 498-535 nm detector. (B) Max projection of the confocal stack obtained from 566-652 nm detector. (C) Image with only GFP signal obtained by subtracting (A) from (B). (D) Merged images of (B) and (C). (E) 3-D reconstruction of (C). Scale bar = 100 µm. Please click here to view a larger version of this figure.
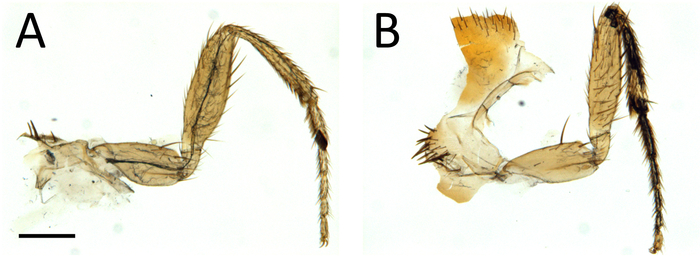
Figure 5: Low-power view of dissected legs showing examples of good (A) and bad (B) fixation. Scale bar = 200 µm. Please click here to view a larger version of this figure.
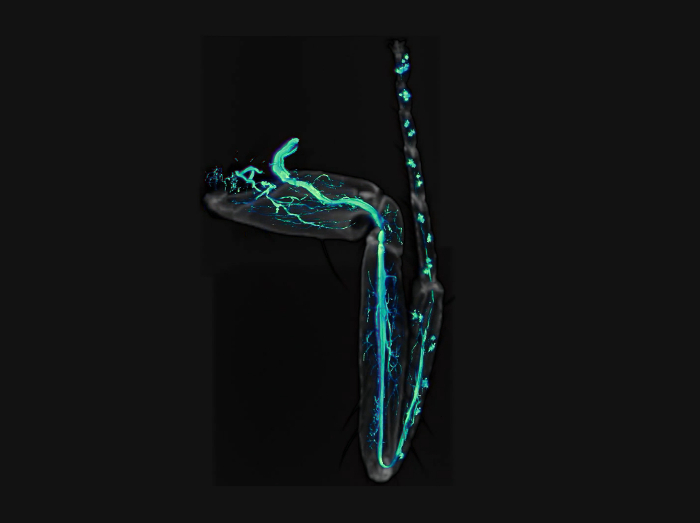
Video 1: Movie of GFP-labeled axons (green) together with cuticle (gray) of a leg. Please click here to view this video. (Right-click to download.)
