We here describe the protocol for how combining TEM imaging together with two electrochemical methodologies, carbon fiber amperometry and intracellular electrochemical cytometry, can provide information that gains a broader view alluding to the effect of extracellular osmotic pressure on secretory vesicles and the exocytosis process. By comparing representative amperometric recordings of exocytosis release at single chromaffin cells using the experimental set up (shown in Figure 1), a significant reduction in exocytotic activity was displayed when cells were exposed to osmotic stress as compared to cells in isotonic conditions (Figure 2A)24. From these recordings and by using Faraday´s law, the total charge detected by each individual amperometric current spike was used to calculate the number of molecules expelled from single vesicle exocytosis events at cells exposed to the different osmotic conditions. By comparison, as displayed in Figure 2B by the smaller area of the average amperometric current spike detected by cells in hypertonic solution, fewer neurotransmitter molecules were released from each vesicle by cells sensing osmotic stress24.
To determine the reversibility of this decline in the number of neurotransmitter molecules released, we performed amperometric recordings of exocytosis at cells exposed to a hypertonic environment and, subsequently, at cells after placed back into an isotonic environment. In these experiments, chromaffin cells were stimulated three consecutive times with BaCl2 solution: first in an isotonic buffer, followed by a second stimulation after cells were incubated for 10 min in a hypertonic solution and, finally, a third stimulation after 10 min cell incubation in isotonic conditions. The results as displayed in Figure 3A present that the quantity of neurotransmitters released was reduced by ~50% when cells were exposed to the hypertonic condition compared to the first Ba2+ stimulation in the isotonic condition. Subsequently, when cells were brought back to an isotonic environment and subjected to a third Ba2+ stimulation, the amount neurotransmitter released per exocytosis event was reversed back to the original amount recorded at the first stimulation, which confirmed previous observations14. Control experiments with three successive Ba2+ stimulations of cells in the isotonic condition (see Figure 3B) show that multiple sequential Ba2+ stimulations in isotonic conditions did not alter the amount neurotransmitter released during exocytosis. This suggests that vesicle quantal size is quickly and reversibly adjusted with extracellular osmolarity.
However, when analyzing the amperometric traces from these experiments in terms of exocytosis activity, it became evident, as displayed in Figure 4A, that exocytosis activity was significantly hampered when cells were exposed to hypertonic stress. During osmotic stress, exocytosis events were reduced to 12% of the activity at cells in isotonic condition. Subsequently, after the osmotic shock and cells were placed back into an isosmotic environment, cells regained 41% of their original exocytosis activity. Interestingly, the control experiments performed in isotonic conditions showed, as displayed in Figure 4B, that after performing three consecutive BaCl2 stimulations, the frequency of exocytosis events was reduced to 53% after a second stimulation and further down to 26% by the third stimulation compared to the first stimulation. Hence, it is clear that consecutive Ba2+ stimulations do not seem to affect the number of neurotransmitters released from each vesicle when exocytosis is triggered, but is significantly influencing the efficiency of the vesicle release process.
To investigate how secretory vesicles in their native environment are affected by extracellular osmotic stress in terms of vesicle volume or quantal size, vesicle size analysis using TEM imaging was combined with intracellular electrochemical cytometry at cells exposed to isotonic and hypertonic conditions. In the intracellular electrochemical cytometry experiments, a carbon fiber nanotip electrode was inserted into the cytoplasm of live chromaffin cells when placed in isotonic and hypertonic solution (as shown in Figure 5.) The resulting amperometric current spike was monitored of each vesicle in the cell cytoplasm colliding, adsorbing, and stochastically rupturing and releasing the vesicle contents at the surface of the amperometric electrode upon bursting26. The integrated total charge detected for each peak in the current versus time trace was used to calculate the average vesicle quantal size in each cell recording by Faraday´s law. These intracellular electrochemical cytometry measurements, shown in Figure 6 and Figure 7B, demonstrated that vesicle quantal size at cells exposed to osmotic stress were significantly reduced compared to at cells in isotonic conditions. Comparing the magnitude of alteration in vesicle quantal size as measured by intracellular electrochemical cytometry, to the fractional decline in the quantity of neurotransmitter released during exocytosis at cells experiencing extracellular osmotic stress, showed a relative decrease of 60% in both quantal size and the amount neurotransmitter released compared to at cells in isotonic conditions (Figure 7)24. To relate the adjustment in vesicle quantal size to a potential variation in vesicle neurotransmitter concentration of cells experiencing osmotic pressure, TEM image analysis was performed to determine the vesicle size of cells exposed to isotonic and hypertonic conditions. Furthermore, the dark staining of the dense core protein matrix inside the LDCVs that is visualized in the TEM images as a dark sphere in the membrane bound vesicles was used to measure the volume of dense core matrix in these vesicles. As presented in Figure 8, vesicle size was reduced to 60% at cells exposed to extracellular osmotic stress compared to at cells in isotonic conditions. From the calculated volume of the measured diameter of the LDCV and the dense core, the volume of the surrounding halo solution that appears as a clear solution inside the LDCVs in the TEM images was also calculated. The summarized results from the TEM image analysis showed, as displayed in Figure 8, that during extracellular osmotic shock it is mainly the volume of the halo solution in the LDCVs that is reduced24.
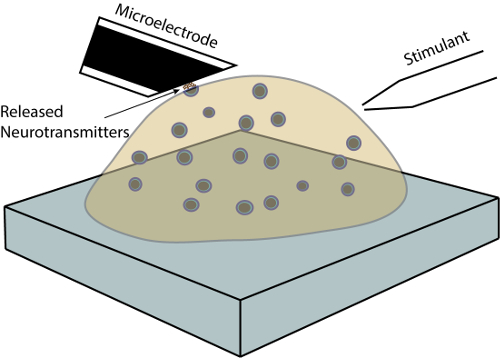
Figure 1: Single cell exocytosis amperometry. A schematic of the experimental set up for amperometric measurement of exocytosis at single chromaffin cells24. Please click here to view a larger version of this figure.
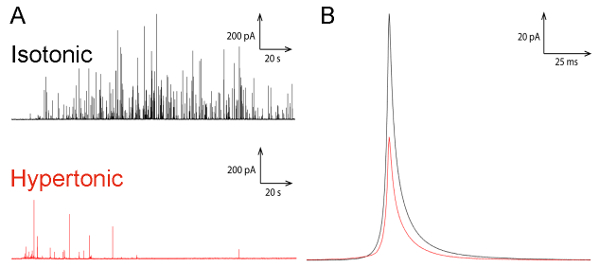
Figure 2: Amperometric traces from exocytosis measurements. (A) A representative amperometric recording of exocytosis at chromaffin cells in isotonic (black color) and in hypertonic (red color) extracellular environments. (B) Enlargement of an average amperometric spike from exocytosis measurement of chromaffin cells in isotonic (black) and hypertonic (red) extracellular environments24. Please click here to view a larger version of this figure.
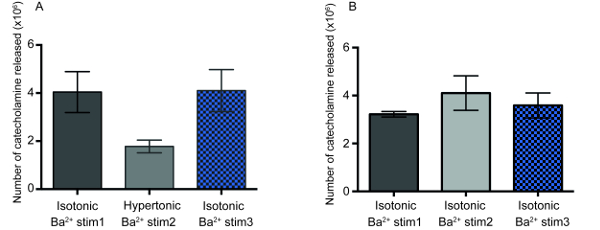
Figure 3: The reversibility of the number of catecholamines released at exocytosis after osmotic shock. (A) The number of molecules released during exocytosis from chromaffin cells (n=4) by three consecutive Ba2+ stimulations, with the first stimulation in isotonic, the second in hypertonic, and the third in isotonic buffer. The statistical results of the unpaired t-test are shown. The p-value for comparison of the first Ba2+ stimulation (Ba2+ stim 1) in isotonic buffer with the second Ba2+ stimulation (Ba2+ stim 2) in hypertonic buffer is p=0.1088 and the p-value for the comparison of the second Ba2+ stimulation in hypertonic solution with the third Ba2+ stimulation (Ba2+ stim 3) in isotonic buffer is p=0.059. (B) Control experiment demonstrating the amount of neurotransmitter molecules released at three consecutive Ba2+ stimulations of chromaffin cells (n= 4) in isotonic buffer. Please click here to view a larger version of this figure.
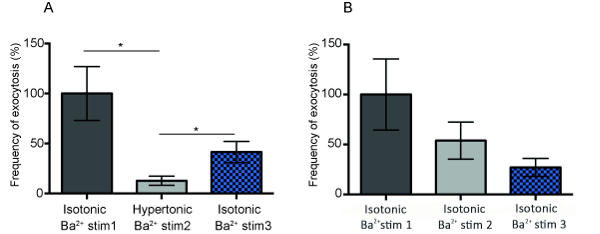
Figure 4: The effect of osmotic pressure on exocytosis activity. (A) The effect of extracellular osmolarity on exocytosis activity is presented as the frequency of exocytosis events when chromaffin cells (n= 4) are stimulated with barium solution in isotonic, then hypertonic, and finally in isotonic conditions. The values are presented as the average number of spikes from each cell and the average from all cells sampled ( standard error of the mean (SEM)). The statistical significance of changes is presented using a t-test for unpaired data (the p-value for isotonic Ba2+ stimulation 1 and hypertonic Ba2+ stimulation 2 is p=0.0126 and the p-value for comparison of hypertonic Ba2+ stimulation 2 and isotonic Ba2+ stimulation 3 is p=0.037).(B) Control experiment presenting the frequency of exocytosis events after three consecutive barium stimulations at chromaffin cells (n=4) in isotonic conditions. Please click here to view a larger version of this figure.
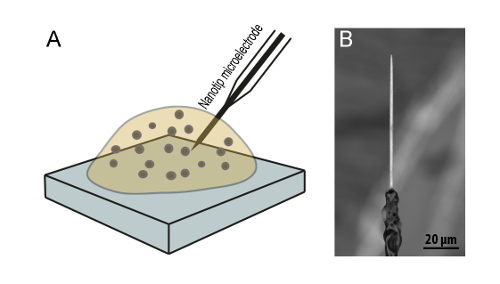
Figure 5: Intracellular vesicle electrochemical cytometry to monitor changes in vesicle quantal size. (A) A schematic of the experimental set up used for intracellular electrochemical cytometry. (B) A scanning electron microscopy image of a typical nanotip conical carbon fiber electrode24. Please click here to view a larger version of this figure.

Figure 6: Intracellular electrochemical cytometry measurements showing (A) Representative traces of intracellular amperometric cytometry recordings at chromaffin cells in isotonic (black) and hypertonic (red) extracellular environments. (B) Enlargement of an average amperometric spike from intracellular cytometry measurement at chromaffin cells in isotonic (black) and hypertonic (red) extracellular environments24. Please click here to view a larger version of this figure.
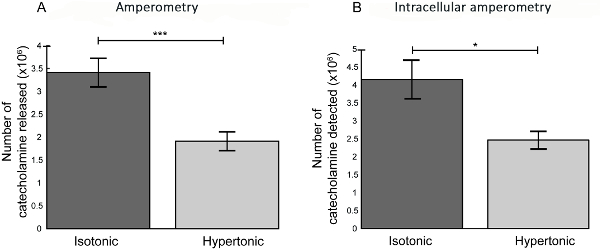
Figure 7: Quantification of the number of catecholamines released at exocytosis and determination of vesicle quantal size. (A) The average number of molecules released during exocytosis recordings at chromaffin cells in isotonic (n=22) and hypertonic (n= 20) environments. The values are presented as an average number of molecules released per exocytosis event from each cell and averaged from the cells sampled ( ± SEM). The statistical significance of changes are presented using t-test for unpaired data (p-value=0.0003) (B) The average number of molecules per vesicle as detected by intracellular electrochemical cytometry at chromaffin cells in isotonic (n=19) and hypertonic (n=16) conditions. The values are presented as the average number of molecules per spike as detected from each cell and averaged from all cells sampled ( ± SEM). The statistical significance of changes is presented using t-test for unpaired data (p- value=0.0108)24. Please click here to view a larger version of this figure.
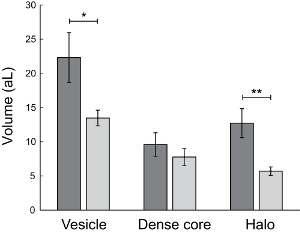
Figure 8: Effect of osmotic pressure on large dense core vesicle size. The calculated volume of the LDCVs, the dense core protein and the halo solution surrounding the dense core protein matrix was calculated in attolitres (aL) from image analysis of TEM images of chromaffin cells in isotonic (n=12) and hypertonic (n=9) buffers. The results were collected from an average of 311 vesicles per cell and averages from single cells ( ± SEM). The p-values are reported from unpaired t-tests comparing isotonic and hypertonic buffers. The P-value is 0.0385 (*) for vesicle volume, 0.3967 for dense core volume, 0.0047 (**) for halo volume24. Please click here to view a larger version of this figure.
