1. Sample Preparation
- Sample preparation (adherent cells; MCF-7 cells)
- Take out the cell culture dish from the incubator and drain the culture medium.
- Rinse the cells on a dish with 1x phosphate-buffered saline (PBS) to remove excessive culture medium.
- Add 3 mL of a solution of 0.25% trypsin to the culture dish (diameter of 100 mm) and put it in a 37 °C, 5% CO2 incubator for 4 min.
NOTE: The trypsin dissolves the adhesive protein of the cells so that they will detach from the culture dish. - Check to see if all the cells have detached from the culture dish using a light microscope (10X objective). If not, gently shake the cell culture dish.
- Add 4 mL of the standard culture medium (formulated with 89% Dulbecco's Modified Eagle Medium (DMEM), 10% fetal bovine serum (FBS), and 1% penicillin-streptomycin (PS)) to stop the action of the trypsin.
- Transfer the whole mixture (cells in trypsin solution and culture medium) to a centrifuge tube and centrifuge for 5 min at 200 x g.
- Remove all the fluid and re-suspend the cells in 1 mL of 1x PBS (pH value: 7.4, pre-heated to 37 °C).
- Sample preparation (cultured micro-algae)
- Transfer the cultured micro-algae and the culture medium (e.g., sea water, agar, or fresh water) to new glass culture tubes (~15 mL) in a volume ratio (micro-algae to culture medium) of 1:5 to establish a new sub-culture medium.
- Place the glass culture tubes in a dust-proof chamber under constant illumination by artificial lighting from fluorescent bulbs according to a light/dark (LD) cycle (usually LD 16:8) for 72 – 120 h before the experiment.
- Transfer the sub-cultured samples to a centrifuge tube and mix well.
2. ATOM System Setup
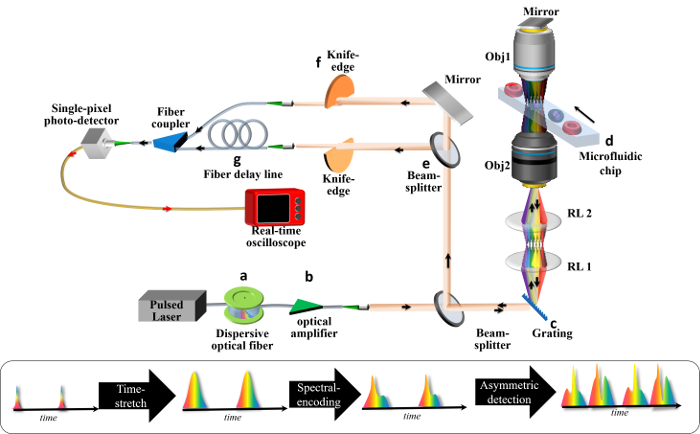
Figure 1: Schematic of an ATOM System. A broadband pulsed laser is employed to deliver ultrafast pulses to (a) a time-stretch module and (b) an in-line optical amplifier module. The time-stretch module generates a train of temporal waveforms, each of which is the replica of the wavelength spectrum of the laser source, (i.e., wavelength-to-time mapping). The amplifier module is used to for pulse-stretching (dispersive) loss compensation. The stretched pulse is then (c) spatially dispersed by a diffraction grating, forming a 1D spectral shower illumination in which individual wavelength components are relayed by a relay lens pair and focused by the objective lens onto different positions on the flowing cell inside the (d) microfluidic chip. This is the process of spectral-encoding. The spectrally encoded light will again pass the cell through another objective lens and a mirror, returning to the diffraction grating and recombining as a spatially undispersed pulsed beam. This image-encoded pulsed beam is then (e) split into two paths, such that both beams are partially blocked with (f) the knife edge, but from opposite directions, before being coupled into the fibers. These two beams represent the two (opposite) encoded phase-gradient contrasts of the final image. For the simultaneous detection of both signal contrasts, one of the signals undergoes (g) a time-delay line, such that the two signals are multiplexed (interleaved) in time. A high-speed photodetector and real-time oscilloscope are used for data acquisition. Please click here to view a larger version of this figure.
- Time stretch module
- Employ a broadband femtosecond (fs) or picosecond (ps) pulsed laser source with a recommended center wavelength in the near infrared (NIR) range, 800-1,500 nm.
NOTE: The typical required pulse width can range from sub-100 fs to a few ps. Detailed requirements of the pulsed laser sources can be seen in References 4 and 5. Important metrics are highlighted in Table 1.- Ensure that the laser source has a high repetition rate, which should be in the megahertz regime (e.g., tens of MHz) to ensure ultrafast imaging in ATOM. Also, set the peak output laser power well below the damage power threshold of the fiber cavity, approximately 1 kW.
- Ensure good shot-to-shot temporal and spectral stability of the pulsed laser source.
NOTE: Typical tolerance in the spectral amplitude fluctuation should be kept within 1.2%19,20,21, as achieved by the fiber mode-locked laser in this setup.
NOTE: The optical bandwidth of the laser source is expected to be 10-100 nm, which is essential to guarantee sufficient imaging field-of-view (FOV) in single-cell imaging21.
- Through a fiber collimator, couple the laser pulsed beam to a single-mode dispersive optical fiber, in which the pulses are stretched via group velocity dispersion (GVD) (Figure 1a).
NOTE: This is the process after which the spectrum of each pulse is mapped onto time as a wavelength-swept waveform (i.e., wavelength-to-time mapping).
NOTE: The total required GVD should be sufficient to ensure that the overall ATOM image resolution is not affected by the wavelength-to-time mapping process (See the Discussion). Typically, in the NIR range, the GVD should be well beyond 0.1 ns/nm. For instance, a single-mode fiber employed in this current setup provides a total GVD of 0.38 ns/nm around the wavelength of 1,060 nm (a total fiber length of 10 km).
- Employ a broadband femtosecond (fs) or picosecond (ps) pulsed laser source with a recommended center wavelength in the near infrared (NIR) range, 800-1,500 nm.
- Spectral-encoding module
- Construct an optical microscope system to perform the spectrally encoded imaging of the cells flowing along the microfluidic channel, as illustrated in Figure 1.
NOTE: The key components of this optical microscope include: (1) a diffraction grating, a telescopic relay-lens module (RL1 and RL2 in Figure 1), and two objective lenses (Obj1 and Obj2 in Figure 1).- First, illuminate the time-stretched and collimated beam onto a diffraction grating (transmission-type grating is employed in this setup) to generate a spectral shower (Figure 1c).
NOTE: The power of the diffracted beam (i.e., diffraction efficiency) can be maximized by adjusting the grating orientation closed to the Littrow configuration. - Configure the two relay lenses (RL1 and RL2) in a 4-f imaging system (i.e., place the diffraction grating on the focal plane of RL1 and separate RL1 and RL2 by a distance equal to the sum of their focal lengths. The spectral shower will then be imaged onto the back focal plane of the objective lens Obj1).
- Carefully align the spectral shower to fill the back aperture of the Obj1, such that the spectral shower can be projected and focused onto the image plane of the microscope.
NOTE: Here, the NA of the objective lens (Obj1 in Figure 1) is 0.75. - Place another objective lens (Obj2 in Figure 1) with a similar NA and a plane mirror at the back aperture of this objective lens (Obj2), such that the spectral shower beam can be aligned to double-pass the image plane and return to the diffraction grating.
- Adjust the pair of objective lenses (Obj1 and Obj2) such that their focal planes overlap with each other. Check to see if the spectral shower double-passes the image plane at the same location and returns to the grating following the same path. If not, perform further alignment and tuning of the optical system.
NOTE: The returned light should pass through an additional beam-splitter, such that the light can be transmitted to an asymmetric detection module. - Adjust the optical amplifier gain at a suitable level, such that the resultant signal can be detected by the photodetector with a good SNR, which is typically at >10 dB.
- Place and adjust the position of the microfluidic chip on the sample platform and make sure that the spectral shower, and thus the imaging area, is placed across the microfluidic channel (Figure 1d).
NOTE: The spectral shower is illuminated orthogonally to the flow direction of fluidics inside the microfluidic chip, such that the flowing motion automatically performs two-dimensional (2D) scans.
- First, illuminate the time-stretched and collimated beam onto a diffraction grating (transmission-type grating is employed in this setup) to generate a spectral shower (Figure 1c).
- Construct an optical microscope system to perform the spectrally encoded imaging of the cells flowing along the microfluidic channel, as illustrated in Figure 1.
- Asymmetric detection module
- Place an additional beam-splitter to separate the imaged-encoded beam into two (Figure 1e).
NOTE: Each beam replica is partially blocked by a knife edge. - Measure and record the optical power of the beam before the beam block. Then, manually position the knife edges (mounted on a linear translation stage) such that they roughly block half of the beam (by visual inspection). Afterward, use the optical power meter to monitor the beam power while fine-tuning the position of the knife edges by translating them across the beam.
NOTE: It is suggested that the optimal position is where the power is decreased by ~50% of the original value (i.e., the unblocked case). This is the condition that provides the best combination of image signal strength and image contrast enhancement. - Repeat step 2.3.2 for another beam replica. Note that the orientation of the partial beam block for one beam should be opposite with respect to the other beam (Figure 1f).
- Couple the two partially-blocked beams into two separate, single-mode fiber arms through two-fiber collimators.
NOTE: One of the arms has an extra length of fiber, serving as a fiber-based delay-line, to introduce a time-delay with respect to the other replica (without the delay-line) (Figure 1g). Both are directed to a single fiber by a fiber coupler before photodetection. The time-delay should be long enough to temporally separate the two replicas and short enough to avoid temporal overlap with the next waveform (i.e., the two replicas are always time-interleaved and time-multiplexed in a single fiber prior to detection (Figure 2a)). - Capture and digitize the detected optical signals with the real-time oscilloscope. Note that the bandwidth and thus the sampling rate of the oscilloscope should be sufficiently high to ensure that they do not influence the final image resolution (See the Discussion).
NOTE: Here, with the GVD of 0.38 ns/nm, a backend acquisition bandwidth and sampling rate of >20 GHz and >40 GSa/s, respectively, are required.
- Place an additional beam-splitter to separate the imaged-encoded beam into two (Figure 1e).
3. Experimental Procedures
- Specimen loading
- Perform cell counting under a conventional phase contrast microscope in a standard hemocytometer.
- Adjust the cell density by diluting with 1x PBS (with spring water for micro-algae) and mix well with a pipette.
NOTE: The suggested concentration is from 105 to 106 cells/mL. - Mount the microfluidic chip onto the sample platform of the optical imaging system.
NOTE: The microfluidic chip is primarily made of polydimethylsiloxane (PDMS) and is fabricated using the standard replica molding method. The microfluidic channel is designed with an asymmetric curved channel to generate the inertial flow focusing effect, such that the cells can flow in single file in the imaging section (with a dimension of 80 µm x 80 µm (height x width)) at high speed. - Transfer the density-adjusted cell solution to a 10 mL syringe.
- Connect the syringe to the inlet of the microfluidic chip and a centrifugal tube to the outlet of the microfluidic chip for disposal collection.
- Mount the syringe onto a syringe pump and set a suitable flow rate to give desirable throughput and to avoid imposing excessive shear force between the cells and the channel.
NOTE: The linear speed of the samples should be within 0.5 and 10 m/s. Note that before the actual image recording, it is often necessary to further fine-tune the system, in terms of maximizing the image signal strength and optimizing the image focusing by repeating steps 2.2.1.4-2.2.1.6.
4. Data Acquisition
- Set a suitable number of data points to be saved for each experiment. The number of data points to be saved depends on the size and flow rate of the samples.
NOTE: Typically, it is set to 8-16 million sample points under a sampling rate of 80 GSa/s. - Acquire and save the data in a batch mode using the oscilloscope.
5. Backend Processing
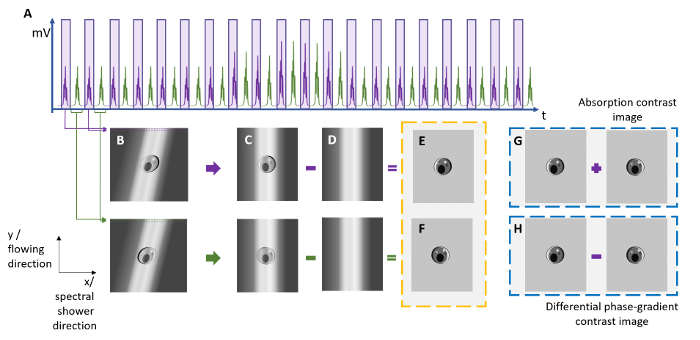
Figure 2: Reconstruction of ATOM Images from the Line-scan Time Trace. To reconstruct the ATOM images with the absorption contrast and differential (enhanced) phase-gradient contrast, two sets of time-stretched pulses (see the purple and green pulses) are extracted from (a) the time-multiplexed temporal waveform trace and are then digitally segmented and stacked to form (b) two 2D images showing the two opposite phase-gradient contrasts. Note that a shearing operation is needed to form (c) the distortion-free images by compensating for the sub-index shift due to the round-off error in the laser repetition rate estimation. By subtracting the (d) raw line-scan from the spectral intensity envelope of the laser source, the background of the images can be removed. (g) An absorption contrast image can be obtained by a pixel-by-pixel intensity summation of images (e) and (f), whereas (h) a differential phase-gradient contrast image can be obtained from the subtraction of images (e) and (f). Please click here to view a larger version of this figure.
- Transfer the data from the oscilloscope to the computers for offline image reconstruction.
NOTE: Here, the data are stored in a portable hard disk and transferred manually between the oscilloscope and the processing computers. The capacity of the hard disk should be more than 500 GB for each experiment.- Use the key image reconstruction routine to digitally stack the line-scans to form a 2D image (Figure 2).
NOTE: The custom program (in MATLAB) then produces two asymmetric detection images by separating the two time-multiplexed data sets (purple and green waveforms in Figure 2).- Obtain a differential phase-gradient contrast image by subtracting the intensities of the two asymmetric detection images; an absorption contrast image can be obtained by adding the two asymmetric detection images. See selected images of MCF-7 and micro-algae in Figure 3. Note that the background profiles of individual images should first be eliminated, and their intensities should be normalized, prior to the image summation and subtraction operations.
- Generate a library of parameters derived from the images, such as cell volume, circularity, and absorption density, etc. for further analysis.
- Use the key image reconstruction routine to digitally stack the line-scans to form a 2D image (Figure 2).
- Input the library of data into the data visualization platform (Figure 4).
- Load the library of parameters and the corresponding reconstructed images to the visualization interface platform.
- Set the axes to be the parameters of interest.
NOTE: The parameters are problem-specific and are defined by the user. They are derived from the ATOM images by the MATLAB program. Here, the optical absorption density of the cell, cell area, cell volume, and cell circularity are available for selection. - Select the dataset to be displayed, such that every cell image can be visualized as a different data point on the scatter plot.
- Move the mouse cursor to each point, such that the corresponding image and other parameters are shown in a floating sub-window.
NOTE: The axes can be changed between the linear and logarithmic scales in an interactive manner. - Perform further analysis of any subset of the dataset by manually gating on the scatter plot.
NOTE: The histogram in every parameter of the gated subset can be plotted against those of the entire library. The separate histograms are displayed in another floating sub-window.
This work illustrates two single-cell imaging demonstrations by ATOM: one with mammalian cells (human peripheral blood mononuclear cells (PBMC) and breast cancer cells (MCF-7)) and another with phytoplanktons (Scenedesmus and Chlamydomonas). The first experiment was motivated by the growing interest in liquid biopsy for the detection, enumeration, and characterization of circulating tumor cells (CTCs) in the blood23. The ability to measure CTCs disseminated from primary tumor sites to the blood stream allows for the examination of metastatic cancer progression24,25. However, challenges exist in CTC analysis: (i) the CTC count is significantly lower than the exorbitant amount of blood cells in whole blood. Current methods of identifying and recovering the CTCs mostly involve cell enrichment and separation steps26,27,28. However, the contaminating background from blood cells and/or the viability of the enriched CTCs remain a concern for subsequent molecular analysis (e.g., RNA sequencing)29. (ii) CTCs are highly heterogeneous in terms of both biophysical and biomolecular signatures, limiting the efficiency of the CTC enrichment and detection. To this end, label-free imaging flow cytometry with a high imaging throughput is an appealing tool that allows for the direct imaging and identification of CTCs within an enormous population of blood cells, minimizing or even bypassing the enrichment steps.
The second experiment is particularly relevant to large-scale phytoplankton characterization, which can impact advances in environmental sciences (e.g., the detection of harmful algal bloom (HAB) species29 and the identification of microalgal species as renewable biodiesel sources)17. Enabled by ATOM, the capability of high-throughput and high-contrast single-cell imaging of phytoplankton could be valuable to reveal the complex heterogeneity in size, texture, and morphology across different genus and species.
Figure 3 shows images of mammalian cells, MCF-7 and PBMC (flowing at a speed of ~10 m/s), as well as microalgae, Scenedesmus and Chlamydomonas (flowing at a speed of ~2 m/s), captured by ATOM. In all four cases, the two opposite phase-gradient contrasts were obtained by the two time-multiplexed, asymmetrically detected signals (described in step 2.3). They both exhibit a pseudo-three-dimensional appearance, resembling the DIC images, each of which shows two opposite shadowing orientations (columns 1 and 2 in Figure 3a-d). Evaluating the pixel-wise intensity sum and difference of these two opposite-contrast ATOM images, it was possible to simultaneously obtain two additional ATOM contrasts (i.e., the absorption-only and enhanced differential phase-gradient contrasts; columns 3 and 4 in Figure 3a-d). Note that not only can label-free ATOM reveal the blur-free single-cell images, but also characterize cellular structures with high contrast. Notably, the vacuoles, pyrenoid, and flagella in the micro-algae can be clearly visualized in Figure 3. This imaging attribute critically enables the extraction of a rich set of identifiers derived from the intrinsic cellular and subcellular textures/morphologies for automated image-based classification.
This work demonstrates that biophysical properties, such as cell circularity and cell size, can be used for the classification of MCF-7 and PBMC. Note that two clusters are observed in the MCF-7 specimen. The availability of high-contrast images for all data points allow us to scrutinize that one of the clusters corresponds to fragments/debris (Figure 4a). A similar classification between two species of phytoplankton based on ATOM images was also performed. Here, the classification results are presented in a form of a 2D scatter plot visualized by the customized user interface (Figure 4). Each annotated data point in the scatter plot, corresponding to each cell, can be further explored with different parameters derived from ATOM. The corresponding ATOM image can also be interactively displayed directly on the plot. Manual gating is also supported in this visualization interface to facilitate further classification and analysis.
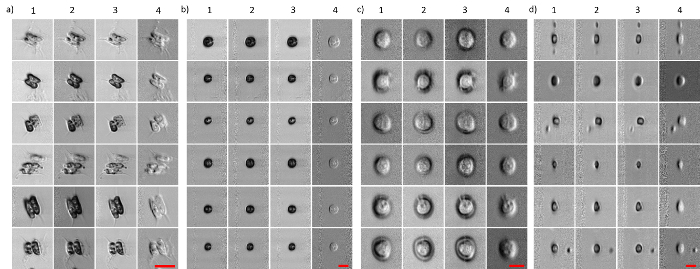
Figure 3: ATOM Image Galley of Micro-algae and Mammalian Cells at an Ultrafast Flowing Speed (~2-10 m/s). (a) Scenedesmus; (b) Chlamydomonas; (c) breast cancer cells, MCF-7; and (d) human PBMC. The Scenedesmus and Chlamydomonas are flowed at ~2 m/s, whereas MCF-7 and PBMC are flowed at 10 m/s. The left two columns (columns 1 and 2) for each cell type represent two opposite phase-gradient contrasts. Columns 3 and 4 represent the absorption contrast and differential (enhanced) phase-gradient contrast, respectively. Scale bars = 20 µm (in red). Please click here to view a larger version of this figure.
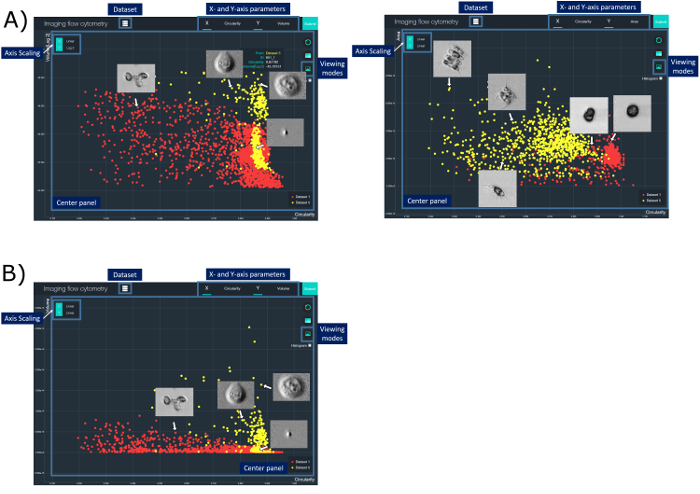
Figure 4: A Graphical User Interface for Cytometric Analysis Based on ATOM Images. In this interface, the user can perform cell-type classification and analysis by defining different cell parameters (e.g., cell size, circularity, absorption density, etc.) derived from the ATOM images. The result can be represented in a scatter plot displayed in the center panel. Access and control of various display options, which include changing parameters and scaling for the x- and y-axis (X- and Y-axis parameter buttons) and switching between data sets (Dataset button), is possible. Detailed cell information (Cell information tab) can be read by hovering on a specific data point. To enable rapid navigation, the user can change to another viewing mode and simultaneously display all images on the scatterplot in the center panel (Viewing modes button). Manual gating can also be performed for further analysis. Yellow data points represent the MCF-7 cell specimen (in (a)) and Scenedesmus (SCE) (in (b)), whereas the red data points represent the human PBMC specimen (in (a)) and Chlamydomonas (CHA) (in (b)). Please click here to view a larger version of this figure.
