Here, we used two models of EAE to understand if a pharmacological agent provides CNS protection by either attenuating CNS-infiltrating T cells or preventing myelin and axonal injury during the onslaught of inflammatory immune cell infiltration. To determine if a therapeutic agent prevents immune cell infiltration into the spinal cord, the C57BL/6 mouse model of chronic EAE is used where immune cell infiltration and disease pathology is predominantly located in the spinal cord (Figure 1A). To determine if a therapeutic drug provides CNS protection during the intrusion of immune cells into the CNS, the SJL animal model of relapsing-remitting EAE is used, which demonstrates disease pathology in both the brain and spinal cord (Figure 1B).
Clinical Assessments
Relevant clinical assessments are made according to the following rubric for typical (Figure 1C) or atypical (Figure 1D) EAE. For typical clinical disease, a score of 0 is no abnormal behavior. When picked up by the base of the tail, the tail may rotate quickly (much like a helicopter rotor) and the hind legs will spread apart. A clinical score of 1 is a partially limp tail, which may be determined by lifting the mouse by the base of the tail. The normal helicopter-like rotating may be weakened or absent, and part of the tail may be completely limp. A helpful way to determine extent of tail paralysis is to run one's finger up the length of the tail, as an unparalyzed tail will usually curl around the finger while a partially paralyzed tail will be unable to do so. A clinical score of 2 represents a completely paralyzed tail. No movement of the tail occurs at all when picking the mouse up at the base of the tail. A clinical score of 3 represents partial hind limb paralysis. Determination of this score requires that the mouse be free to move on a flat surface. If one hind limb is dragging as the mouse moves forward, or if one or both hind limbs appear to be partially paralyzed, a score of 3 may be given. A clinical score of 4 represents complete hind limb paralysis. With this score, a mouse will be unable to move its hind limbs and will drag itself forward using its front limbs. A clinical score of 5 represents a moribund mouse, or a mouse with difficulty moving itself across its cage or breathing. If a mouse cannot drag itself along the bottom of the cage or if its breathing is labored, the mouse should be humanely euthanized. A clinical score of 6 represents a mouse found dead in its cage. A score of 6 is unusual and causes of death other than EAE should be investigated.
Atypical clinical disease may or may not be accompanied by paralysis. It may be necessary to include two separate scoring systems if a mouse presents with atypical disease plus typical symptoms. A score of 0 is no abnormal behavior, as with the typical scoring system. A clinical score of 1 represents a slight head turn or tilt while the mouse is walking. This may be determined by allowing the mouse to walk forward and observing a constant left or right directionality to its movement. A clinical score of 2 represents a more pronounced head turn and poor righting ability. As with an atypical score of 1, the mouse has directionality to its movement and may have slight difficulty with balance. A clinical score of 3 represents an inability to walk in a straight line. The mouse will have difficulty balancing and may use the side of the cage to help right itself as it walks. A clinical score of 4 represents a mouse laying on its side, unable to walk due to balancing issues. The mouse may be able to drag itself along the bottom of the cage but may have directionality to its movement. A clinical score of 5 represents continuous rolling unless supported. A mouse that reaches this score should be humanely euthanized. A clinical score of 6 represents a mouse found dead in its cage. A score of 6 is unusual and causes of death other than EAE should be investigated.
It may be necessary to allow for "in-between" scores, e.g., adding 0.5 to a score if a mouse's condition changes slightly or if choosing between two scores is difficult. For example, a mouse that begins to move more slowly than its normal counterparts but displays no paralysis, or a mouse that clasps its hind feet with its front instead of splaying its legs out when picked up by the tail may be given a score of 0.5. A mouse that can only drag itself along the bottom of the cage and is only able to twitch its hind limbs periodically or when touched may be given a score of 3.5.
Assessing a Reduction in Immune Cell Infiltration
After induction of EAE in the C57BL/6 mouse model (Figure 1A, day 0), antigen presentation and proliferation of T cells in the spleen occur on days 1 – 5 followed by immune cell infiltration into the CNS around day 7. Approximately 3 to 5 days after the initial immune cell infiltration mice present with clinical scores. To assess if a therapeutic agent is blocking immune cell infiltration into the spinal cord, drugs or vehicle are introduced on day 7 after antigen presentation and proliferation in the spleens but before immune cells start to infiltrate into the spinal cord. If immune cell infiltration has been attenuated, the clinical disease course should reflect improved clinical scores during the rising phase of the disease from days 10 to 15 (Figure 2).
A reduction in immune cell infiltration would also result in diminished neuroinflammation. Reactive astrocytosis and microgliosis are considered major hallmarks for neuroinflammation. Staining for astrocytes with GFAP and microglia with Iba-1 can then be used to assess changes in mean area fraction staining to quantify neuroinflammation (Figure 3).
To determine if immune cell infiltration is reduced, the spinal cords are removed and processed for flow cytometry analysis at the peak of disease (Figure 1A, approximately day 18). This ensures that the largest number of immune cells have entered into the spinal cord. Entrance of T cells into the CNS is considered the initiating inflammatory event and both Th1 and Th17 cells are found in animal models of EAE as well as MS patients. Taken together, flow cytometric analysis should include assessment of both types of pathogenic T cells. Furthermore, Tregs are well-characterized suppressor T cells that dampen disease. Therefore, the percentage of Tregs from a total CD4+ population must be evaluated compared to the percentage of effector T cell populations. This will reveal if an overall reduction in T cell infiltration has occurred or if there is a skewing of T cell phenotypes in the CNS. Representative dot plots (Figure 4A) demonstrate a reduction in overall number of CD4+ infiltrating T cells in spinal cords from drug-treated mice compared with spinal cords from vehicle-treated mice (numbers in upper right quadrants). To evaluate Th1, Th17, and Treg cells the following signature proteins are evaluated: IFN-γ+, IL-17+, and Foxp3+, respectively and should be reduced (Figure 4A). Statistical analysis should be performed on CD4+, IFN-γ+, IL-17+, and Foxp3+ cell numbers to demonstrate a significant reduction (Figure 4B). To rule out a skewing of T cell subsets, statistical evaluation of the proportion of IFN-γ+ IL-17+, IFN-γ+ IL-17–, IL-17+ IFN-γ–, and Foxp3+ cells is performed (Figure 4C).
To eliminate the possibility that a reduction in CNS-infiltrating T cells is a consequence of inhibiting proliferation, activation, and differentiation in the periphery, the number of actively proliferating T cells in addition to the proportion of T cell subtypes needs to be evaluated. No change in the percentage of CD4+, IFN-γ+, IL-17+, or Foxp3+ should be found if activation and differentiation are unaffected (Figure 5A). Furthermore, no change in Ki67+ CD4+ cells should be found if proliferation is unaffected (Figure 5B). Drug treatments are introduced on day 7 or later to avoid altering initial antigen presentation and T cell activation in the periphery. However, in genetic models proteins are often deleted constitutively during embryogenesis or induced before induction of EAE making splenocyte assessment of high importance.
Assessing CNS Protection
To demonstrate if a particular therapeutic agent modulates disease pathology in the CNS after immune cell infiltration, drug interventions should be administered during the first peak in clinical disease scoring. The SJL model of EAE is advantageous for these experiments since these mice exhibit a relapsing-remitting phenotype. If a drug treatment prevents myelin-axon degeneration, an improvement in clinical scores will be observed (Figure 6). Pathological assessment of myelin must corroborate a reduction in myelin damage consistent with improved clinical scores. To quantitatively evaluate myelin integrity, DAB staining of myelin basic protein (MBP) is performed, followed by statistical analysis of the optical density for this staining (Figure 7). To further substantiate that neuroinflammation is sustained or decreased by therapeutic interventions, reactive gliosis can be assessed by measuring mean fraction area for reactive gliosis as described above (Figure 3). To corroborate that a therapeutic intervention is directly protecting the CNS without immunomodulatory effects, attenuation of immune cell infiltration into the CNS and proliferation in the spleens must be discounted. To address this, methods for brain and spinal cord assessment of immune cell infiltration and assessment of peripheral T cell proliferation and activation should be performed as described above (Figures 4 and 5). Taken together, therapeutic agents that block cell injury in the CNS with no evidence of a reduction in CNS-infiltrating T cells or proliferation of T cells in the periphery are CNS-protective treatments.
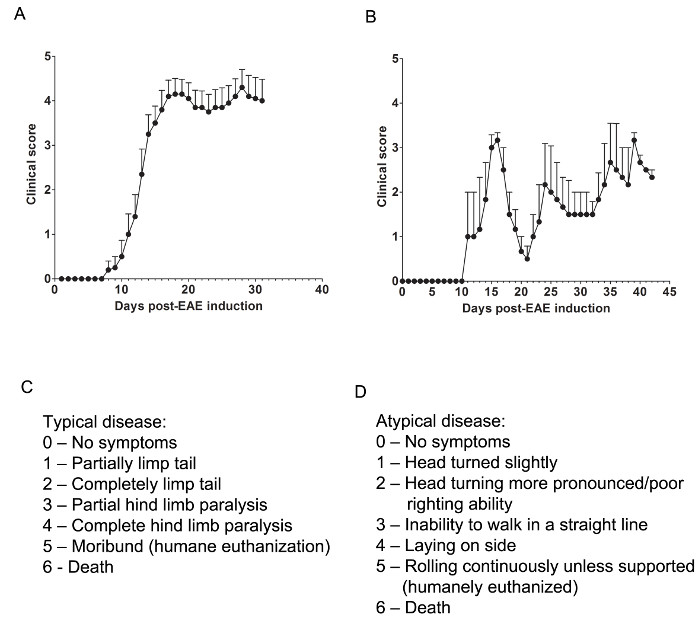
Figure 1. Representative Results of Clinical Scores from EAE in C57BL/6 and SJL Mice. (A) Clinical scores (mean ± SEM) of C57BL/6 mice (n = 10) induced with MOG35-55 to produce EAE with chronic disease. (B) Clinical scores (mean ± SEM) of SJL mice (n = 3) induced with PLP139-151 to produce EAE with relapsing-remitting disease. (C) The clinical scoring rubric used to track typical disease progression in EAE mice. (D) The clinical scoring rubric used to track atypical disease progression in EAE mice. Please click here to view a larger version of this figure.
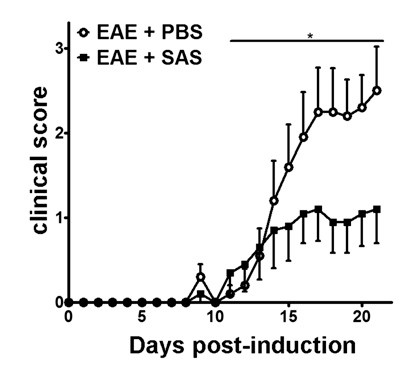
Figure 2. Pharmacological Treatment prior to Immune Cell Infiltration in C57BL/6 mice with EAE. Clinical scores (mean ± SEM) of C57BL/6 mice treated with PBS (n = 20) or SAS (n = 19) from day 7 postimmunization with MOG35-55. Data are from three pooled independent experiments. Statistical difference was determined using a nonparametric two-tailed Mann-Whitney U test, *p < 0.05. Re-print with permission from (11).
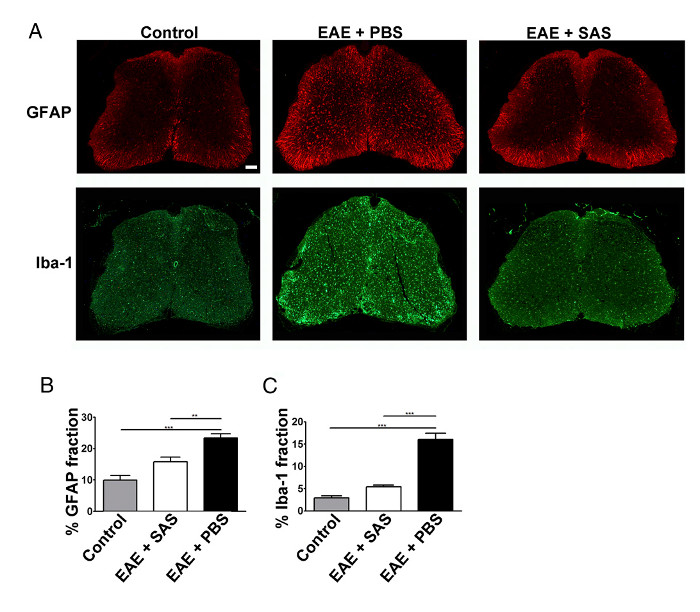
Figure 3. Immunofluorescent Staining and Quantification of Reactive Gliosis in Spinal Cords of Control, EAE, and Treated C57BL/6 Mice. (A) Fluorescent labeling for GFAP (astrocytes) and Iba-1 (microglia) in the spinal cords of control (unimmunized) mice (left panels) and EAE mice treated with PBS (middle panels) or SAS (right panels). Scale bar = 100 µm. Quantification of staining was determined using the area fraction technique to measure percent immunopositive area for GFAP (B) and Iba-1 (C). Mean ± SEM, n = 3 control, n = 3 SAS-treated, or n = 4 PBS-treated mice, 6 sections per mouse. Statistical differences were determined using a one-way ANOVA, *p < 0.05, **p < 0.01, ***p < 0.001. Re-print with permission from (11). Please click here to view a larger version of this figure.
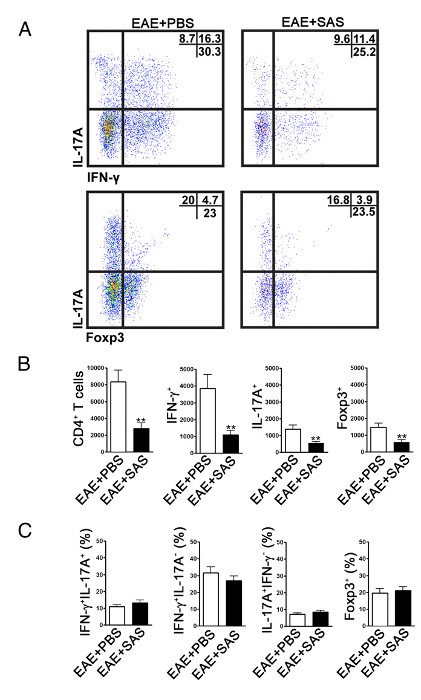
Figure 4. FACS Analysis of EAE C57BL/6 Mouse Spinal Cords Demonstrating Reduced T cell Infiltration in Treated Mice. C57BL/6 mice were treated with SAS or PBS, beginning 7 d postinduction of EAE. Spinal cords were obtained on day 15. (A) Representative dot plots show Th1 (IFN-γ+/IL-17–) and Th17 (IFN-γ–/IL-17+) cells in CD4+ gate (upper panels) and T regulatory cells (Foxp3+) (lower panels). Dot plots show percentages in upper right quadrant. (B) Absolute numbers of CD4+ cells as well as IFN-γ+, IL-17A+, and Foxp3+ cells were statistically analyzed. (C) The change in percentage of T cell populations between SAS- and PBS-treated EAE mice was also examined. Mean ± SEM, n = 10 for PBS treated, and n = 9 for SAS treated from two independent experiments. Two-tailed t test was used for all bar graphs. **p < 0.01. Re-print with permission from (11). Please click here to view a larger version of this figure.
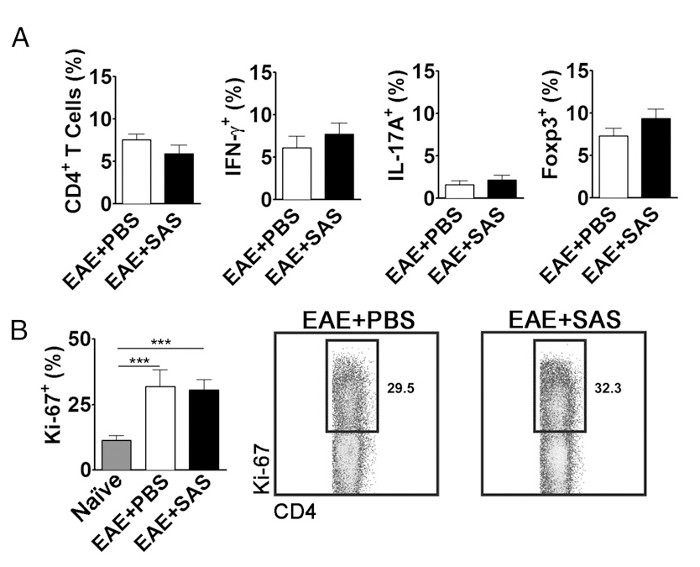
Figure 5. FACS Analysis of EAE C57BL/6 Mouse Spleens Demonstrating Equivalent T cell Expression Profiles and Proliferation in Treated and Untreated Mice. Spleens from PBS- and SAS-treated mice were analyzed 15 d postinduction of EAE. (A) The percentage of CD4+ T cells, Th1 (IFN-γ+/IL-17–), Th17 (IFN-γ–/IL-17+), and T regulatory cells (Foxp3+) in spleens from PBS-treated (n = 10) and SAS-treated (n = 9) mice from two independent experiments. (B, left panel) The percentage of Ki-67+ cells in the CD4+ population from naive spleens (n = 4) as well as from PBS- (n = 5) and SAS-treated mice (n = 5) induced with EAE. A one-way ANOVA test demonstrated statistical significance between the proportion of Ki-67+ cells from naive spleens compared with either PBS- or SAS-treated EAE spleens. No significance was observed between PBS- and SAS-treated EAE spleens. (B, right panel) Representative dot plots; numbers indicate proportion of proliferation. Dot plots show percentages. Bar graphs represent two-tailed t test, ***p < 0.001. Re-print with permission from (11). Please click here to view a larger version of this figure.
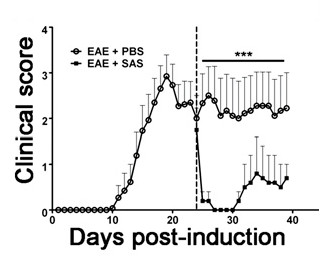
Figure 6. Pharmacological Treatment after Immune Cell Infiltration in SJL Mice with EAE. Clinical scores (mean ± SEM) of SJL mice treated with PBS (n = 8) or SAS (n = 8) from day 24 postimmunization (dashed line) with PLP139-151. Data are mean ± SEM of clinical scores. Statistical difference was determined using a nonparametric two-tailed Mann-Whitney U test, ***p < 0.001. Top line represents values used for statistical analysis. Re-print with permission from (11).
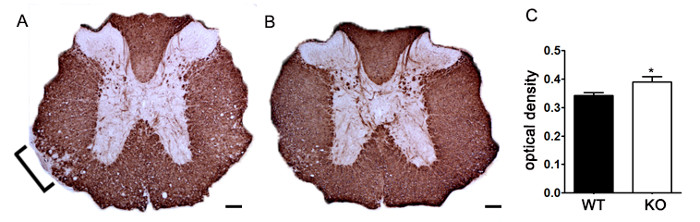
Figure 7. Quantification of MBP Staining using Optical Density. (A) Representative staining of MBP in thoracic spinal cord from an unspecified genetic knockout mouse compared to littermate control C57BL/6 mouse induced with EAE. Bracket indicates representative area of reduced MBP staining indicating demyelination. (B) MBP staining of thoracic spinal cord from an unspecified genetic knockout C57BL/6 mouse. (C) Unspecified genetic knockout mice induced with EAE (KO; n = 6 mice, 2 – 4 lumbar and thoracic sections per animal) exhibit a higher optical density (OD) of MBP staining in the spinal cord than wildtype (WT; n = 3 mice, 2 – 4 lumbar and thoracic sections per animal) mice induced with EAE. Statistically analyzed using a two-tailed t test, *p < 0.05. Error bars represent SEM. Scale bar 100 µm.
