1. Preparation of culture media – all reagents are listed in Table 1
- Thaw male rat serum at 37°C.
- Heat-Inactivate rat serum for 30 mins at 55°C
- Centrifuge rat serum at 10K for 5 mins at room temperature
- Remove supernatant and mix 50:50 with phenol-red free DMEM w/ High Glucose
- Sterilize the media using a 0.45 um syringe filter
- At least 1ml of media / embryo should be prepared
2. Isolation of the uterus from the pregnant dam
- Using procedures approved by the Institutional Animal Care and Use Committee (IACUC), the pregnant dam is first anesthetized and then sacrificed by cervical dislocation
- Sterilize the abdomen with 70% EtOH
- Pinch a small path of skin along the midline, just above the nipple line, with large forceps and open the abdomen underneath your forceps with large scissors. Be careful not to damage any internal structures
- Use your scissors to cut laterally from the initial opening toward each side of the mouse so that the entire abdomen is open
- Intestines and excess fat pads can often obscure the uterus and these should be moved aside so that the top of the uterus and ovaries can be observed on each side
- Pinch the top of the uterus below the ovaries and use your scissors to separate the uterus from the fat, cutting from one end of the uterus to the other ovary
- Lift out the uterus with your forceps and place in a Petri dish with saline
3. Removal of embryos from the uterus
- Rinse the uterus with saline to remove any excess blood and trim any excess fat from the outside
- Transfer to a clean dish with room temp. Tyrodes saline to improve visualization
- Using a stereomicroscope, separate each embryo using either small scissors or if necessary fine (#5) forceps to carefully peel the uterus apart.
- Insert fine forceps into the space between the uterine wall and the decidua and peel back the uterine wall, being careful not to pierce the embryo
4. Removal of decidua from embryos
- Orient the decidua so that the darker portion (placenta) is facing away from you
- Make an incision with your forceps along the line separating the dark portion of the decidua from the lighter portion, being careful not to cut too deeply
- Complete the separation of the placenta from the top of the yolk sac, being careful not to tear the ectoplacental cone (EPC). At this point you should be able to visualize the Reicherts membrane on the yolk sac
- Continue to remove the entire decidua (lighter portion) being careful not to pierce the yolk sac
- Gently pinch and remove the Reicherts by peeling it away from the EPC. If the EPC is damaged or separated from the yolk sac, embryos will fail to turn normally from E8.5-E9.5
- With the Reicherts removed the embryo should be easily visible through the yolk sac and can be staged by counting somities.
5. Setting up the culture system
- Cut the end of a plastic transfer pipette with scissors to increase the diameter of the opening so that an embryo can be pipetted without damaging the yolk sac
- Fill clean roller bottles with 1 ml of media per embryo. Depending on the type of roller bottles used a maximum of 3-6 embryos can be loaded in each bottle.
- Transfer the embryos using your cut pipette to the media-filled roller bottle, carrying over as little Tyrodes as possible so as not to dilute the media
- Attach the rubber plug (bung) to the top of the glass roller bottle
- Insert the roller bottles into the roller culture drum so that a tight seal is formed between the plug and the drum
- Once all the bottles have been attached, and any empty slots in the drum have been sealed with rubber stoppers, activate the roller
- Open CO2 and O2 tanks and adjust them to approximately 2 psi, so that the outlet valve is releasing one bubble per second. For E8.5-E10 embryos, 20% O2 and 5% CO2 should be used.
- Ensure that the incubator is set to 37°C and cover or close the incubator so that the embryos are shielded from light.
- Embryos should be checked periodically to assess their development by the addition of somities, the progression of turning, and the extent of neural tube closure
- After 2-3 hours in culture, pharmacological inhibitors or other treatments can be added to the media
- The study design should include controls, such as vehicle or inactive analogues of study reagent.
6. Evaluating development after whole embryo culture
- Turn off gas, stop roller, and remove bottles from incubator
- Transfer embryos back to Tyrodes or PBS in a Petri dish for evaluation under stereomicroscope
- The yolk sac should appear balloon-like, a heartbeat should be visible and heart rate should be >120 beats per min, and circulation should be evident
- Photograph embryos prior to yolk sac removal if necessary
- Remove yolk sac from embryo by cutting a hole near the EPC and flipping the yolk sac and amnion over the head and rump of the embryo.
- The umbilical cord can be cut to separate the yolk sac from the embryo, and the yolk sac can be used for genotyping the embryo if necessary.
- Examine the embryo for somite number and neural tube defects, such as open cranial folds, incomplete closure of the face and/or an open caudal neuropore. Embryos can be staged according to the Theiler classification of multiple morphogenetic changes (http://www.emouseatlas.org/ emap/home.html) Again, a photographic record of embryo development is useful.
7. Notes
- A high quality dissection is essential, because any nick or tears in the yolk sac will prevent successful turning and inhibit normal development. The EPC can also be damaged during removal of the Reichert’s membrane (RM), and if the EPC is even partially separated from the yolk sac, embryos will not develop normally. However, incomplete removal of the RM can cause embryos to stick together and/or RM cells may overgrow in culture and constrict yolk sac expansion, both of which would prevent normal development.
- Clean and sharp forceps are the secret to getting a good dissection, because dull or bent forceps make the procedure significantly more difficult. Protein deposits on instruments will lead to tissue damage.
- Clean, sterile glass bottles are also essential to prevent embryos from sticking to the side wall and being damaged. Prior to starting the experiment, bottles should be scrubbed with soap and water, rinsed in dH2O, and microwaved with a small amount of water in each bottle until the water boils. Bottles are then rinsed with 70% EtOH and allowed to air dry.
- Work as quickly as possible, because the sooner the embryo can be transferred to the culture media and warmed back to 37°C the better survival.
- Complete dissecting the entire litter before transferring any embryos to roller bottles so that each embryo is at room temperature for the same amount of time to avoid creating any discrepancies in embryo growth
- Maintain a sterile environment to prevent bacterial contamination of the cultures. Our culture experiments have been performed without antibiotic, but many labs add antibiotic to their media to prevent bacterial growth. The medium should appear as clear at the end of the experiment as it was at the beginning of the experiment. If media has become cloudy or there is a visible precipitate, there is likely a contamination that has infected your culture.
- Ineffective gas exchange can also delay embryos development, so be sure to have a reliable regulator that will ensure continuous delivery.
- Embryos develop approximately 50% slower in ex vivo culture, therefore it is essential to count somites to ensure that you have reached the appropriate stage of development for completion of the experiment.
8. Representative results:
The appearance of embryos pre- and post- roller culture is illustrated in Figure 1. At the time of dissection, embryos should be in an unturned configuration (Fig. 1A,D) where the tail is behind the head folds. After 36 hrs in culture, embryos should have completed turning so that they are in the C-curved, fetal position, where the tail is in front of the head (Fig 1B,C,E,F). Pharmacological manipulation with RhoA kinase inhibitor (Y-27632), a known inhibitor of convergent extension during neurulation (Ybot-Gonzalez, 2007), results in a shortening of the embryos along their rostral-caudal axis (Fig. 1E,F) and inhibits cranial neural fold closure. Our data show that increasing doses of Y-27632 progressively impairs cranial fold closure (Figure 2A) and shortens the body axis (Fig. 2B), consistent with the role of downstream RhoA signaling in cranial neurulation and convergent extension.
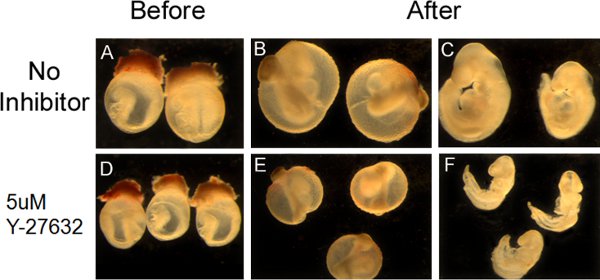
Figure 1. Appearance of cultures embryos and manipulation with the pharmacological inhibitor Y-27632. (A,D) Dissected embryos at E8.5 prior to whole embryo culture (B,E) Embryos at E10 with the yolk sac still intact, subsequent to 36hrs of roller culture. (C,F) The yolk sac has been removed to illustrate successful turning and neural tube closure. Embryos that were treated with the Rho kinase inhibitor Y-27632 (E,F) failed to undergo proper convergent extension and illustrate a shortened body axis.
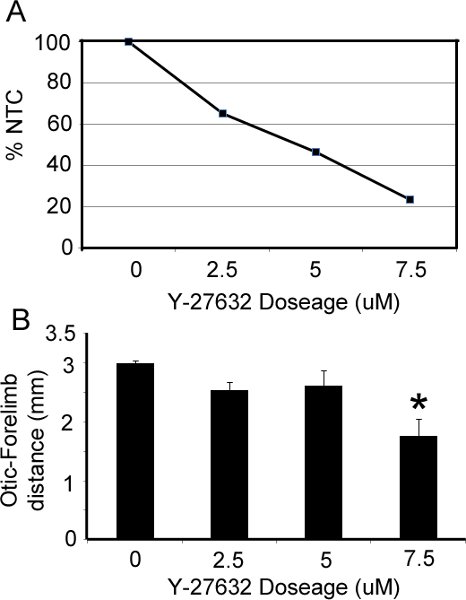
Figure 2. Effect of RhoA kinase inhibitor on cranial neural tube closure and axis elongation. (A) The percentage of embryos that were able to successfully close their cranial folds (%NTC= percentage neural tube closure) is compared to the dose of Y-27632 added to culture media. (B) The distance between the otic vesicle and forelimb was significantly reduced at increasing doses of Y-27632 (p<.05).
