
Figure 1. Views of mounted microfluidic co-culture device (A) from above; (B) partially lateral. The culture chambers (cc) are highlighted in red (eosin) and blue (toluidine blue); microgrooves (mg) can be seen as a white line between the culture chambers. Ganglia and co-cultured organs/tissues are placed into the appropriate culture chamber via the punched holes (ph) in the device. Media are added and removed via the wells (mw).
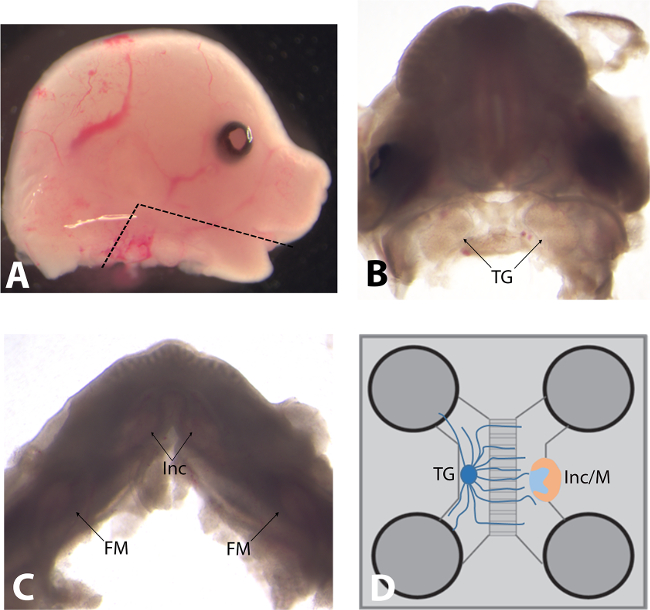
Figure 2. (A) Lateral view of a mouse embryo head (embryonic day of development E14.5). The black line indicates where the lower jaw should be cut in order to preserve the integrity of both trigeminal ganglia and tooth germs. (B) Mouse embryo head (E14.5) after removal of lower jaw, skull, and telencephalon. Black arrows indicate trigeminal ganglia (TG). (C) The lower jaw of mouse embryo (E14.5) after removal of the tongue. Black arrows indicate the localization of the different tooth germs (Inc: incisor; FM: first molars). (D) Schematic representation of the microfluidic co-culture device.
