Photoconversion-Based Leukocyte Tracking in Bacterial-Infected Transgenic Zebrafish
Abstract
Source: Harvie, E. A. et al., Non-invasive Imaging of the Innate Immune Response in a Zebrafish Larval Model of Streptococcus iniae Infection. J. Vis. Exp. (2015)
This video demonstrates the tracking of leukocyte recruitment using transgenic zebrafish larvae expressing photoconvertible green fluorescent protein. Pre-injecting red fluorescent dye-labeled bacteria into the otic vesicle and inducing photoconversion enables observation of leukocytes' migration, phagocytosis, and dispersal throughout the body, providing valuable insights into immune response dynamics.
Protocol
1. Preparing Microinjection Needles
- Prepare thin wall glass capillary injection needles (1.0 OD/0.75 ID) using a micropipette puller device with the following settings: air pressure 200, heat 502, pull 90, velocity 80, time 70, air time at the start of pull 5, air time at end of pull 5.
- Using fine tweezers, break off the tip of the pulled needle so that the tip opening has a diameter of approximately 10 µm.
2. Preparing Larval Injection Dishes
- Rinse a gel comb with a lane width of approximately 4-5 mm in sterile water and allow it to dry.
- Prepare a 1.5-2% high melt agarose solution in E3 medium and microwave until the solution is clear. Once cooled, pour some of the agarose into a Petri dish (100 x 15 mm) and swirl, adding just enough agarose to completely cover the bottom of the dish.
- Once the agarose layer has solidified, place the rinsed and dried gel comb on top so that the non-combed end is just resting on the top of the Petri dish and the combed end is touching the agarose. Ensure that the comb is as horizontal as possible, creating a 30° angle with respect to the bottom agarose layer.
- Pour an additional small amount of agarose at the interface between the comb and bottom agarose layer so that the fresh agarose layer covers the wells of the comb. Allow to cool completely before removing the comb. Using a pipet tip, remove any overhanging pieces of agarose from the wells.
- Pour E3 medium on top of the injection mold and store at 4 °C.
- Before each use, replace with fresh medium and warm injection ramps at 28.5 °C for at least an hour before injection.
- Immediately prior to injections, replace the E3 on the injection ramp with an E3 medium containing 200 µg/ml ethyl 3-aminobenzoate (tricaine).
3. Preparing Streptococcus iniae (S. iniae) Inoculum
- Prepare and autoclave Todd Hewitt broth medium supplemented with 0.2% yeast extract and 2% proteose peptone (THY+P): 30 g/L Todd Hewitt, 2 g/L yeast extract, 20 g/L proteose peptone. For agar plates, add 14 g/L agar.
- Prepare bacterial cultures the night before infections by pipetting a 100 µl aliquot of frozen bacterial stock into 10 ml of THY+P broth in a sealed 15 ml tube. Incubate overnight without agitation at 37 °C. After 14-16 hr of growth, use the overnight culture either to make freezer stocks or prepare for use in injections.
- Make freezer stocks by placing 1 ml of the overnight culture in 500 µl of 80% glycerol in a 1.7 ml centrifuge tube. To avoid freeze-thaw cycles, make 100 µl one-use aliquots from this mix and store them at -80 °C.
- For S. iniae used in infections, dilute the overnight culture 1:100 for a total volume of 10 ml culture by adding 0.1 ml of overnight culture to 9.9 ml of THY+P broth. Grow at 37 °C without agitation for approximately 4-5 hr. Monitor the optical density (OD) at 600 nm using a Nanodrop spectrophotometer and harvest the bacteria in a mid-logarithmic phase when the OD600 nm reaches 0.250-0.500. An OD600 nm of 0.250 corresponds to approximately 108 colony-forming units (CFU)/ml.
- Pellet 1 ml of the bacterial culture in a 1.7 ml centrifuge tube at 1,500 x g for 5 min. Resuspend in 1 ml of fresh phosphate-buffered saline (PBS) and repeat. Measure the OD600 nm of the bacteria in PBS, pellet, and resuspend in PBS to achieve the desired concentration.
- To aid in the visualization of microinjection, add phenol red to the bacterial suspensions prior to injection for a final concentration of 0.1%.
- For experiments involving the injection of heat-killed bacteria, heat the bacteria in PBS at 95 °C for 30 min. Confirm that the heat-killing process reduced the number of viable bacteria to undetectable levels by plating an injection volume (approximately 1 nl) on solid THY+P agar plates and incubating overnight at 37 °C.
4. Labeling S. iniae with a CellTracker Red Fluorescent Dye
- To label living bacterial cells, prepare a stock solution of a CellTracker fluorescent dye or equivalent. As the dye used comes in 20 x 50 µg aliquots of powder and needs to be resuspended in dimethyl sulfoxide (DMSO), add 7.3 µl DMSO to the tube to obtain a 10 mM stock concentration.
- Test a range of dye concentrations (e.g., 0.5-25 µM) on the bacteria to determine the lowest optimal concentration that stains the cells. Rapidly dividing cells and longer experiments may require a higher concentration of dye.
- Pellet 1.0 ml of bacterial culture in a 1.7 ml tube by centrifugation as described above (section 3). Resuspend the pellet in 1 ml fresh PBS and add the appropriate volume of dye to the bacterial culture.
- Incubate without agitation at 37 °C for 30 min. Spin down the bacteria and resuspend in 1 ml of pre-warmed THY+P broth and incubate without agitation for an additional 30 min at 37 °C.
- Spin down the bacteria and wash them two times in PBS before measuring the OD600 nm and diluting the bacteria for microinjection as detailed above (section 3). We typically inject approximately 100 CFU in 1 nl injection volume for our studies of leukocyte recruitment and phagocytosis.
5. Preparation of Zebrafish Larvae for Infections
- Set up breeding pairs the night before and collect embryos as described by Rosen et al. Incubate embryos in E3 medium at 28.5 °C until ready to infect.
- For zebrafish that will be imaged, prevent the development of pigment (melanization) by adding N-Phenylthiourea (PTU) to the E3 medium at 24 hr post fertilization (hpf) for a final concentration of 0.2 nM.
- For infections involving embryos aged 2 days post fertilization (dpf), dechorionate embryos manually with a pair of fine tweezers. Alternatively, dechorionate embryos by removing E3 and replacing them with pronase (2 mg/ml) for about 5 min or until gentle pipetting breaks embryos out of the chorion. Most larvae should have hatched naturally by 3 dpf.
- Anesthetize zebrafish several minutes prior to infection by placing dechorionated larvae into an E3 medium containing 200 µg/ml tricaine.
6. Otic Vesicle Injection of S. iniae into Three-Day-Old Larvae
- Turn on the microinjector and set the time range to "millisecond". Open the valve on the carbon dioxide tank to let gas into the line. The pressure of the microinjector should read approximately 20 pounds per square inch (PSI). Adjust the pressure in the microinjector unit by clockwise turning of the black knurled knob for increased pressure or counterclockwise turning for decreased pressure.
- Vortex the prepared S. iniae culture in phenol red and PBS and use a microloader tip to load 2-3 µl of the culture into a pulled capillary injection needle.
- Mount the loaded needle on a micromanipulator connected to a magnetic stand and position it under a stereomicroscope so that the needle is at approximately a 45-65° angle with respect to the base of the microscope.
- Press on the foot pedal of the microinjector to dispense a drop of the inoculum onto the tip of the needle. Measure the diameter of the drop using the scale bar in the ocular lens of the microscope.
- Alternatively, estimate the diameter of the drop by injecting a volume into a drop of mineral oil on a glass microscope slide with a scale bar. The diameter of the drop should be approximately 0.10 mm, which is about a 1 nl volume.
- Adjust the drop size by adjusting the duration setting on the microinjector or by clipping off more of the needle tip with fine tweezers. Note that the injection time should be between 20 – 35 msec to avoid causing too much tissue damage.
- Using a plastic transfer pipet, transfer 12 anesthetized larvae into each well of the injection mold.
- Use a glass rod, hair loop, or plastic tip to gently position the larvae so that the heads are pointed towards the back of the microscope and the yolk sacs are against the left side of the well. Point the left ear of the larva up towards the ceiling.
- Looking through the ocular lens of the stereomicroscope, use the knobs on the micromanipulator to line up the loaded needle with the otic vesicle so that both are in the same field of view. Pierce the outer epithelial layer of the otic vesicle with the needle tip so that the needle tip is just inside the vesicle.
- Press on the foot pedal to inject 1 nl of the desired dose of S. iniae. Be sure to use a low enough pressure so as not to rupture the cavity. If the injection is successful, the otic vesicle, but not the surrounding tissue, should fill with the phenol red inoculum (Figure 1 Ai). Immediately remove any mis-injected fish from the injection plate.
- Carefully retract the needle out of the larva and move the injection plate by hand so that the next larva is in view. Do this under 5X magnification.
NOTE: Retraction of the needle may result in the deposition of some bacteria outside of the otic vesicle, which may skew survival and leukocyte recruitment results. To avoid this, it is recommended to inject fluorescent dye-labeled bacteria and visually scan injected larvae under a fluorescent microscope to remove any larvae where this has occurred. A correctly injected larva is shown in (Figure 1 Bi). - To ensure the injection volume/inoculum remains the same over the course of the experiment, inject a drop of the bacterial suspension into a 1.7 ml centrifuge tube containing 100 µl of sterile PBS after every 48th embryo. Plate the 100 µl on THY+P agar plates at 37 °C overnight to determine the CFU in the injection volume.
- When the entire group of 12 larvae on the injection ramp has been injected, carefully use a plastic transfer pipette to remove the larvae from the wells and place them into a new Petri dish (35 x 10 mm2). Remove the tricaine solution and replace it with approximately 2 ml of fresh E3 medium to allow the larvae to recover.
- Pipet injected larvae into single wells of a 96-well plate and incubated at 28.5 °C. Monitor larvae over time for survival in these plates or remove them later for imaging or CFU counts.
7. Fixation of Larvae for Imaging
- Fix larvae in an ice-cold 4% paraformaldehyde in PBS solution as described in section 7.
- At room temperature, wash 3 times for 5 min each in PBS before imaging.
8. Preparation of Larvae for Live Imaging
- Prepare a 1.5% low-melting-point agarose solution in E3 by heating it in the microwave until the solution is clear.
- Place agarose solution in a 55 °C water bath to let it cool but not harden.
- Add tricaine to the agarose to a final concentration of 0.016%.
- Using a plastic transfer pipette, place 4-5 anesthetized larvae in a glass bottom dish on the stage of a stereomicroscope.
- Remove the tricaine and agarose solution from the 55 °C water bath and let it cool at room temperature for 1-2 min. While the agarose is cooling, remove as much liquid from the anesthetized larvae as possible.
- Pour the cooled agarose into the dish until about half of the surface is covered. Swirl the dish to spread the agarose. Note that if the agarose is too hot, it will kill the larvae. Also, if too much agarose is added to the dish, the objective lens of the microscope may hit the bottom of the dish when focusing through the agarose.
- Using a transfer pipette, pick up the larvae that have floated to the sides of the dish and pipet them back into the center.
- Under the stereomicroscope, gently position the larvae as desired with a hair loop, a long pipette tip, or a glass rod. For imaging of the otic vesicle, position the larvae so that the left otic vesicle is flat against the bottom of the dish.
- Let the agarose cool for about 10 min before moving the dish. The larvae may shift positions if the agarose is not solid. Gently pipet some tricaine and E3 solution to the top of the agarose layer to keep it moist.
9. Confocal Imaging of Infection
- Place the glass bottom dish with larvae onto the stage of an inverted microscope with an FV-1000 laser scanning confocal system.
- Set the pinhole to 200-300 µm and using a numeric aperture 0.75/20X objective lens, set z-stacks with 3-6 µm slices.
- Use continuous line scanning to adjust the laser power and detector gain for each channel.
- Using sequential line scanning for each fluorescence channel (e.g., 488 and 543 nm) and differential interference contrast (DIC), conduct a time-lapse movie of the left otic vesicle every 3 min for 2-6 hr to observe initial recruitment and phagocytosis by neutrophils and macrophages. For longer time courses, place a lid on the Petri dish to prevent evaporation and drying out of the agarose.
- Acquire still images of fixed (Figure 1B) or live (Figure 2) larvae at 20X or 40X magnification.
10. Photoconversion of Dendra2-labeled Leukocytes at the Otic Vesicle
NOTE: Dendra2 can be photoconverted from green to red fluorescence by focusing a 405 nm laser (50-70% laser power should be sufficient) on the region of interest (ROI) for 1 min. Below is the step-by-step protocol used for the FV-1000 laser scanning confocal system:
- Visualize the sample using a z-stack scan with the 488 nm and 543 nm lasers. Use continuous line scanning to adjust the laser power and detector gain. Check to make sure there is no accidentally photoconverted red fluorescence.
- On the "Image Acquisition Control" window, under "Stimulus Setting" select the "Use Scanner" tool and choose "Main". Select the 405 nm laser and set it at 70% power. Using the circle option, define the ROI in the otic vesicle.
- Under "Stimulus Start Setting" select "Activation in series" with a preactivation of 1 frame and an activation time of 60,000 msec. On the "Acquisition Setting" window under the "Time Scan heading", choose 2 intervals of 00:01:00 (one for pre- and one for post-photoconversion).
NOTE: After the time-lapse series scan is complete, the Dendra2 should have been photoconverted to its red fluorescent state. - Scan the sample by a z-stack using the 488 nm and 543 nm lasers to visualize the photoconverted red fluorescence as well as any remaining green fluorescence (Figure 3).
Representative Results

Figure 1: Leukocyte recruitment to otic vesicle infection with S. iniae. (A) Neutrophil recruitment to S. iniae infection. (i) Successful injection of a phenol red-labeled inoculum into the otic vesicle. (ii–iv) Sudan Black staining of larvae for investigation of neutrophil recruitment at 2 hpi. PBS mock-infected larvae show little recruitment of neutrophils to the otic vesicle (ii) whereas infection with either wild-type S. iniae or the cpsA mutant results in robust neutrophil recruitment (iii, iv). Scale bar, 300 µm. (B) Macrophage recruitment to S. iniae infection. (i) Successful microinjection of red-labeled S. iniae (depicted in magenta) into the otic vesicle. (ii–iv) Fluorescent confocal images of microinjected transgenic mpeg1:dendra2 larvae fixed at 2 hpi. PBS mock-infected larvae show little macrophage recruitment (ii), but larvae infected with CellTracker Red-labeled (depicted in magenta) wild-type S. iniae or the cpsA mutant show robust macrophage recruitment to the otic vesicle at 2 hpi (iii, iv). Scale bar, 30 µm.

Figure 2: Phagocytosis of S. iniae by phagocytes in the otic vesicle. Transgenic mpx:dendra2 (A) or mpeg1:dendra2 (B) larva infected with red-labeled S. iniae (depicted in magenta) and imaged at 60 min post-infection using a laser scanning confocal microscope. Scale bar, 30 µm.
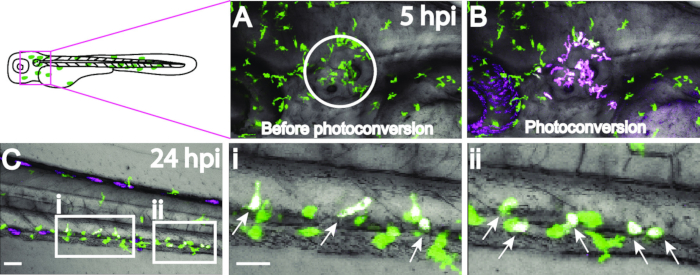
Figure 3: Photoconversion of macrophages at the otic vesicle 5 hpi with S. iniae. Macrophages (depicted in green) at the otic vesicle, designated by the circle (A), were photoconverted (B) using a 405 nm laser on a confocal microscope and tracked over time. By 24 hpi, photoconverted macrophages (depicted in magenta) have migrated as far as the trunk/caudal hematopoietic tissue (C); scale bar, 50 µm. Higher magnifications of the boxed regions in C are shown in (i) and (ii), scale bar 30 µm; arrows point to photoconverted macrophages. Photoconverted cells appear white because of the merged 543 nm red fluorescence and any remaining 488 nm green fluorescence.
Divulgations
The authors have nothing to disclose.
Materials
| 1.7 ml eppendorfs | MidSci | AVSS1700 | |
| 14 ml falcon tube | BD Falcon | 352059 | |
| 96 well plate | Corning Incorporated | 3596 | |
| Agar | BD Biosciences | 214030 | |
| CellTracker Red | Molecular Probes, Invitrogen | C34552 | |
| CNA agar | Dot Scientific, Inc | 7126A | |
| Disposable transfer pipets | Fisher Scientific | 13-711-7m | |
| Dissecting Scope | Nikon | SMZ745 | |
| DMSO | Sigma Aldrich | D2650 | |
| Fine tweezers | Fine Science Tools | 11251-20 | |
| Gel comb | VWR | 27372-482 | 4.2 mm width, 1.5 mm thick |
| Glass bottom dishes | Custom made by drilling a 16–18 mm hole in the center of a 35-mm tissue culture dish bottom and placing a 22-mm round #1 coverslip in the hole and sealing with a thin layer of Norland Optical Adhesive 68 cured by UV light. | ||
| Glycerol | Fisher Scientific | G33-4 | |
| High melt agarose | Denville Scientific, Inc. | CA3510-6 | |
| Laser Scanning Confocal Microscope | Olympus | with FV-1000 system | |
| Low melt agarose | Fisher | BP165-25 | |
| Magnetic stand | Tritech (Narishige) | GJ-1 | |
| Microinjection system | Parker | Picospritzer III | |
| Microloader pipet tips | Eppendorf | 930001007 | |
| Micromanipulator | Tritech (Narishige) | M-152 | |
| Micropipette puller | Sutter Instrument Company | Flaming/Brown P-97 | |
| Nanodrop spectrophotmeter | Thermo Scientific | ND-1000 | |
| N-Phenylthiourea (PTU) | Sigma aldrich | P7629 | |
| Paraformaldheyde | Electron Microscopy Sciences | 15710 | |
| Petri Dishes | Fisher Scientific | FB0875712 | 100 mm x 15 mm |
| Phenol Red | Ricca Chemoical Company | 572516 | |
| Phosphate Buffered Saline | Fisher Scientific | BP665-1 | |
| Pronase | Roche | 165921 | |
| Protease peptone | Fluka Biochemika | 29185 | |
| Small cell culture dish | Corning Incorporated | 430165 | 35 mm x 10 mm |
| Thin wall glass capillary injection needles | World Precision Instruments, Inc. | TW100-3 | |
| Todd Hewitt | Sigma Aldrich/Fluka Analytical | T1438 | |
| Tricaine (ethyl 3-aminobenzoate) | Argent Chemical Laboratory/Finquel | C-FINQ-UE-100G | |
| Yeast extract | Fluka Biochemika | 92144 |
