The details of all the synthesized products (1-5), including the structure, full NMR spectra, optical purity, and HRMS-MALDI data, are provided in Supplementary File 1.
1. Synthesis of 3-(aziridin-2-yl)acryl aldehyde (1a)
- Flame dry a 50 mL round-bottomed flask equipped with a stirrer bar and a septum under vacuum conditions. Cool it to room temperature while filling it with argon gas.
- Add anhydrous toluene (19 mL) and (R)-1-((R)-1-phenylethyl)aziridine-2-carbaldehyde (1.00 g, 5.71 mmol) (see Table of Materials) to the flask. Then, stir the solution for 1 min.
- Add (triphenylphosphoranylidene)acetaldehyde (2.08 g, 6.85 mmol) (see Table of Materials) to the stirred solution.
- Heat the reaction mixture at 60 °C for 18 h. Then, cool the reaction mixture to room temperature and remove volatile solvent from the reaction mixture under reduced pressure.
- Monitor the reaction progress by TLC using ethyl acetate:hexanes (1:6 v/v, Rf = 0.25) as an eluent.
- Purify the crude product by silica gel flash chromatography with ethyl acetate:hexanes (1:6 v/v) as the eluent to isolate pure product 1a (see Supplementary File 1) as a yellow liquid.
- Confirm the product by NMR and polarimeter measurements (see steps 8 and 9 for measurement methods).
2. Synthesis of contiguous bisaziridine (2a)
- Flame dry a 50 mL round-bottomed flask with a stirrer bar and a septum under vacuum conditions. Cool it to room temperature while filling it with argon gas.
- Add anhydrous ethyl acetate (3 mL) and 1a (step 1.6, 201 mg, 1.0 mmol) to the flask, and then stir the solution for 1 min.
- Add catalyst (S)-2-(diphenyl((trimethylsilyl)oxy)methyl)pyrrolidine (BS, see Table of Materials) (0.02 mL, 7 mol%) to the mixture and stir at room temperature for 30 min.
- Add 316 mg, 1.10 mmol of tert-butyl tosyloxycarbamate (BocNHOTs, see Table of Materials) and 123 mg, 1.50 mmol of sodium acetate to the reaction mixture and stir for 24 h.
- Monitor the reaction progress by TLC using diethyl ether:hexanes (1:4 v/v, Rf = 0.27) as an eluent.
- Extract the reaction mixture with ethyl acetate (3 x 50 mL) in a separatory funnel.
- Dry the combined organic layer over anhydrous Na2SO4, filter, and concentrate in vacuo.
- Purify the resulting crude product by flash chromatography on silica gel with diethyl ether:hexanes (1:4 v/v) as eluent to isolate pure product 2a (see Supplementary File 1) as a yellow liquid.
- Confirm the product by NMR and polarimeter measurements (see steps 8 and 9).
3. Synthesis of compound 3
- Flame dry a 50 mL round-bottomed flask with a stirrer bar and a septum under vacuum conditions. Cool it to room temperature while filling it with argon gas.
- Add anhydrous methanol (11 mL) and aldehyde 2a (step 2.8, 1.00 g, 3.16 mmol) [or 2b (1.17 g, 3.16 mmol, see Supplementary File 1)] to the flask, and then stir the solution for a minute.
- Add NaBH4 (95 mg, 2.53 mmol) to the stirred solution.
- Stir the reaction mixture at 0 °C for 1 h.
- Monitor the reaction progress by TLC using ethyl acetate:hexanes (1:4 v/v, Rf = 0.27) as an eluent.
- After 1 h, quench the reaction mixture with distilled water and extract with ethyl acetate (3 x 50 mL) in a separatory funnel.
- Dry the combined organic layer over anhydrous Na2SO4, filter, and concentrate in vacuo.
- Purify the crude residue by silica gel flash chromatography with ethyl acetate:hexanes (1:4 v/v) as the eluent to isolate pure products 3a [or 3b] (see Supplementary File 1) as a yellow liquid.
- Confirm the product by NMR and polarimeter measurements (see steps 8 and 9).
4. Synthesis of compound 4
- Flame dry a 50 mL round-bottomed flask with a stirrer bar and a septum under vacuum conditions. Cool it to room temperature while filling it with argon gas.
- Add anhydrous dichloromethane (11 mL) and alcohol 3a (step 3.8, 1.00 g, 3.14 mmol) [or 3b (1.17 g, 3.14 mmol)] to the flask, and then stir the solution for a minute.
- Add tert-butyldimethylsilyl chloride (TBSCl, 520 mg, 3.45 mmol) and imidazole (427 mg, 6.28 mmol) (see Table of Materials) to the stirred solution.
- Stir the reaction mixture at 0 °C for 18 h.
- Monitor the reaction progress by TLC using ethyl acetate:hexanes (1:4 v/v, Rf = 0.26) as an eluent.
- After 18 h, quench the reaction mixture with deionized water and extract with methylene chloride (3 x 50 mL) in a separatory funnel.
- Dry the combined organic layer over the anhydrous sodium sulfate, filter, and then concentrate under reduced pressure.
- Purify the crude residue by silica gel flash chromatography with ethyl acetate:hexanes (1:4 v/v) as the eluent to isolate pure products 4a [or 4b] (see Supplementary File 1) as a yellow liquid.
- Confirm the product by NMR and polarimeter measurements.
5. Selective ring-opening of non-activated aziridines: Synthesis of 5d
- Flame dry a 50 mL round-bottomed flask with a stirrer bar and a septum under vacuum conditions. Cool it to room temperature while filling it with argon gas.
- Add 3b (step 4.2, 100 mg, 0.27 mmol) and acetic acid (0.12 mL, 2.14 mmol) to the flask, and then stir the mixture at room temperature for 5 h.
- Monitor the reaction progress by TLC using ethyl acetate:hexanes (2:3 v/v, Rf = 0.28) as an eluent.
- After 5 h, remove the acetic acid in vacuo.
- Purify the crude residue by silica gel flash chromatography with ethyl acetate:hexanes (2:3 v/v) as the eluent to isolate pure product 5d (see Supplementary File 1) as a yellow liquid.
- Confirm the product by NMR and polarimeter measurements.
6. Selective ring-opening of activated aziridines: Synthesis of 5f
- Flame dry a 50 mL round-bottomed flask with a stirrer bar and a septum under vacuum conditions. Cool it to room temperature while filling it with argon gas.
- Add anhydrous methanol (8 mL) and 4b (step 4.8, 100 mg, 0.21 mmol) to the flask, and then stir the solution for 1 min.
- Add NaN3 (39 mg, 0.6 mmol) and NH4Cl (21 mg, 0.41 mmol) in H2O (1 mL) to the above solution.
- Stir the reaction mixture at 0 °C for 4 h.
- Monitor the reaction progress by TLC using ethyl acetate:hexanes (1:4 v/v, Rf = 0.30) as an eluent.
- After 4 h, quench the reaction mixture with H2O and extract with ethyl acetate (3 x 50 mL) in a separatory funnel.
- Dry the combined organic layer over anhydrous Na2SO4, filter, and concentrate in vacuo.
- Purify the crude residue by silica gel flash chromatography with ethyl acetate:hexanes (1:4 v/v) as the eluent to isolate pure product 5f (see Supplementary File 1) as a yellow liquid.
- Confirm the product by NMR and polarimeter measurements.
7. Pd-catalyzed hydrogenation of contiguous aziridines: Synthesis of 5h
- Flame dry a 50 mL round-bottomed flask with a stirrer bar and a septum under vacuum conditions. Cool it to room temperature while filling it with argon gas.
- Add anhydrous methanol (5 mL), 2b (step 3.2, 100 mg, 0.27 mmol), Boc2O (70 mg, 0.32 mmol), and 20% Pd(OH)2/C (37 mg) to the flask.
- Stir the mixture under H2 atmosphere (balloon, 1 atm) at room temperature for 12 h.
- Monitor the reaction progress by TLC using ethyl acetate:hexanes (2:3 v/v, Rf = 0.29) as an eluent.
- Filter the reaction mixture through a commercially available celite pad (see Table of Materials) and wash with methanol.
- Evaporate in vacuo the filtrate and purify the residue by silica gel flash chromatography with ethyl acetate:hexanes (2:3 v/v) as the eluent to isolate pure product 5h (see Supplementary File 1) as a colorless liquid.
- Confirm the product by NMR and polarimeter measurements.
8. Polarimeter analysis
- Prepare an appropriate amount of the sample (~100 mg) to be measured.
- Dissolve the prepared sample in CHCl3 (c 0.05-1.00).
- Transfer the sample solution into the sample chamber, ensuring that there are no air bubbles
[ø = 1.8 mm, l = 10-1 m]. - Load the sample chamber into the polarimeter instrument (see Table of Materials) and check the orientation of the chamber.
- Set 0 as click zero clear in the 'control' section.
- Measure the specific rotation of CHCl3 for the blank.
NOTE: Light source: Na; λ = 589 nm; D.I.T: 5 s; cycle times: 5; cycle interval: 5 s; temperature: 20 °C. - Measure the specific rotation of the sample solution at a constant temperature.
NOTE: Light source: Na; λ = 589 nm; D.I.T: 5 s; cycle times: 5; cycle interval: 5 s; temperature: 20 °C. - Measure the specific rotation of the sample solution three times in the same way to obtain the average value.
- Calculate the specific rotation using the following equation27:
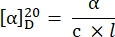
α = observed rotation (degrees), c = concentration (g/mL), l = path length (10-1 m).
9. 1H and 13C NMR analysis
- Prepare approximately 0.6-1.0 mL of the NMR solvent (CDCl3).
- Dissolve ~50 mg of sample in the solvent at a concentration of 0.02 M for 1H NMR and 0.05 M for 13C NMR measurements.
- Transfer the sample solution into an NMR tube using a Pasteur pipette.
- Load the tube into the NMR instrument (see Table of Materials).
NOTE: NMR measurements were performed using 400 MHz or 500 MHz spectrometers. Spin number: 16 (1H NMR), 256 or 512 (13C NMR); measurement time: 10 min (1H NMR), 20 or 30 min (13C NMR)]. - Record the NMR spectra and analyze the data.
NOTE: Reference the chemical shift of spectrum to CDCl3 signal [δ (1H NMR spectrum) = 7.26 ppm; δ (13C NMR spectrum) = 77.0 ppm)].
To investigate the achievability of preparing a contiguous bisaziridine, (E)-3-((S)-1-((R)-1-phenylethyl)aziridin-2-yl)acrylaldehyde (1a) was first synthesized as a model substrate according to the procedure mentioned in step 1 (Figure 1)28.
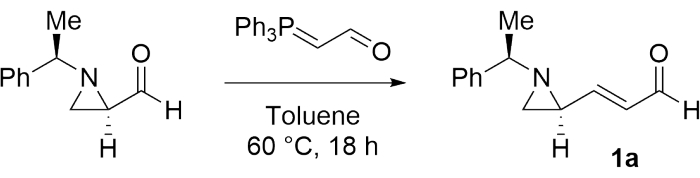
Figure 1: Synthesis of 1a as a model substrate. Product 1a was synthesized from (R)-1-((R)-1-phenylethyl)aziridine-2-carbaldehyde using (triphenylphosphoranylidene)acetaldehyde as the reagent. This figure is adapted from Mao et al.28 and Rhee et al.52. Please click here to view a larger version of this figure.
Thereafter, aziridination of 1a was performed to obtain contiguous bisaziridines (2a and 2b) under the following optimal reaction conditions29,30,31,32,33,34,35 (step 2 and Figure 2): a) For the synthesis of 2a: 1a (1.0 mmol), catalyst BS (7 mol%), BocNHOTs as the nitrogen source (1.1 equivalent), NaOAc as the base (1.5 equivalent), EtOAc (0. 3 M) for 24 h at room temperature; b) For the synthesis of 2b: 1a (1.0 mmol), catalyst BS (7 mol%), TsNHOTs as the nitrogen source (1.1 equivalent), NaOAc as the base (1.5 equivalent), THF (0. 3 M) for 7 h at room temperature.
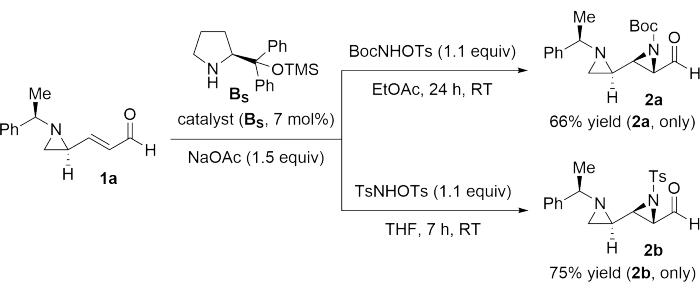
Figure 2: Synthesis of contiguous bisaziridines (2a and 2b). Products 2a and 2b were synthesized from 1a in two steps, using BS as the catalyst in the first step and BocNHOTs or TsNHOTs as the reagent in the second step. Please click here to view a larger version of this figure.
After the construction of enantioenriched bisaziridines bearing non-activated and activated aziridine moieties, various nitrogen-enriched molecules (5a–g) were prepared via regioselective ring-opening reactions with diverse nucleophiles. Representative examples of the ring-opening reactions of bisaziridines are summarized in Table 1.
Table 1: Regioselective ring-opening of bisaziridines with diverse nucleophiles. This table is adapted from Rhee et al.52. Please click here to download this Table.
In the presence of the Lewis acid ZnCl2, the sulfur atom of 1-phenyl-5-mercaptotetrazole and in acidic media NH4Cl, the amine of aniline attacks the less hindered C3 atom of the aziridines 3b and 4b to afford the corresponding products 5a and 5b, respectively36,37,38,39,40,41,42 (Table 1, entries 1 and 2). Product 5c was synthesized when the nitrogen atom of N-methyleneamine equivalent43, which was produced from 1,3,5-triethylhexahydro-1,3,5-triazine in the presence of ZnBr2 as a catalyst, attacks the non-activated aziridine over the unreacted activated aziridine 3b, followed by ring-closure reaction (Table 1, entry 3). Surprisingly, the regiochemical control in the ring-opening of the non-activated aziridines can be achieved by the selection of an appropriate N-protecting group such as Ts or Boc group on the activated aziridines (Table 1, entries 4 and 5). Presumably, the differential ring-opening pattern may be attributed to the geometry of the activation modes upon protonation (i.e., secondary interactions) (Figure 3). The secondary interaction between the nitrogen atom of the non-activated aziridine and the adjacent nitrogen atom of the sulfonamide at the C2' position may have occurred, creating a sterically congested chiral environment; the consequent nucleophilic attack of acetate on the less hindered C3 atom44,45,46 would have led to the formation of the kinetic product 5d (Table 1, entry 4). On the other hand, the secondary interaction between the proton of the aziridinium ion and carbonyl oxygen may involve the construction of a more flexible and less sterically congested circumstance, leading to the formation of thermodynamic product 5e via the nucleophilic attack20,21,22,23,24,25,26 of acetate on the more substituted C2 atom (Table 1, entry 5). Notably, various nucleophiles, such as S, N, C, and O, favorably attacked the non-activated aziridine under mildly acidic conditions (Table 1, entries 1-5).
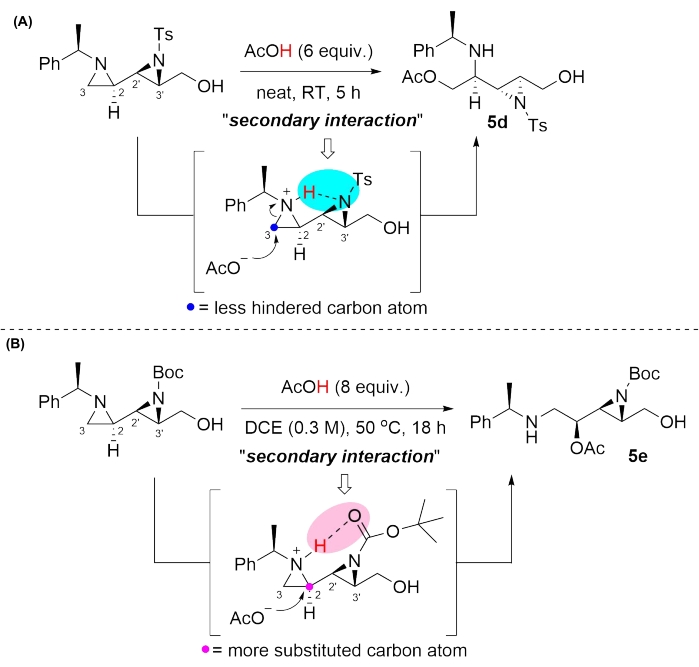
Figure 3: Plausible secondary interactions for the selective ring-opening reactions of contiguous bisaziridines. (A) The nucleophilic attack of acetate on the less hindered C3 atom would have resulted in the formation of kinetic products 5d. (B) The nucleophilic attack of acetate on the more substituted C2 atom would have resulted in the formation of thermodynamic product 5e. This figure is adapted from Rhee et al.52. Please click here to view a larger version of this figure.
The selective ring-opening reaction of N-Ts-protected aziridine can be accomplished with azide (N3ˉ) to afford the desired product 5f, since azide provides access to the less hindered C3' atom47,48,49 (Table 1, entry 6). Further, isoxazoline N-oxide 5g was synthesized via the formation of a β-hydroxy-α-nitro ester and the successive nucleophilic attack of the nitronate oxygen atom on the C3' atom of the activated aziridine ring, while the contiguous bisaziridinyl aldehyde was reacted with ethyl nitroacetate and imidazole50 (Table 1, entry 7). Notably, preferential ring-opening reactions of the activated aziridine moieties occurred under basic conditions (Table 1, entries 6 and 7).
In the presence of Pd(OH)2/C, H2 (1 atm) and Boc2O, the contiguous bisaziridinyl aldehyde was easily converted to the multi-substituted chiral pyrrolidine compound 5h via the following consecutive reactions51 (Table 1, entry 8 and Figure 4).
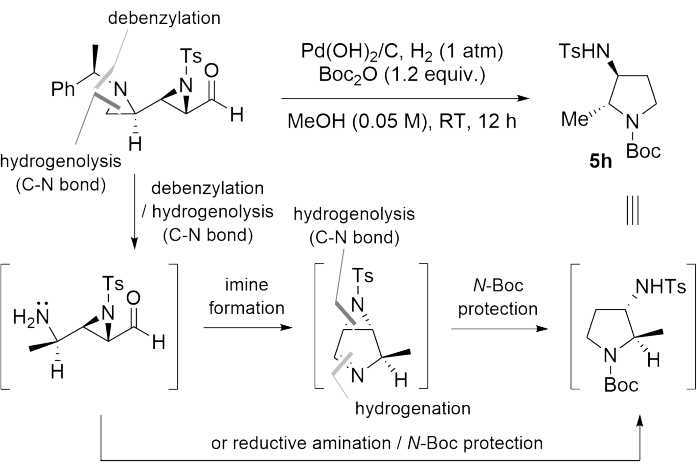
Figure 4: Schematic illustration for the synthesis of chiral pyrrolidine. This figure has been modified from Rhee et al.52. Please click here to view a larger version of this figure.
Characterization data for the products
Some important peaks in the 1H NMR spectral data (Figure 5, Figure 6, Figure 7, Figure 8, Figure 9, Figure 10, and Figure 11) of the compounds are as follows. The peak of the aldehyde hydrogen appears at ≥9.00 ppm. The peaks of the alkene hydrogens appear in the range of 5.00-7.00 ppm. The peaks of the aziridine hydrogens appear at ≤3.50 ppm. In the case of bisaziridine, the hydrogens appear individually. Generally, the peaks of the hydrogens of the other alkyl groups appear at ≤3.00 ppm. In the case of Boc and TBS, the hydrogen peaks are generally stationary and appear as singlets at ≤2.00 ppm. In the case of the bisaziridine ring-opening compound, the hydrogen peaks of the alkyl group appear individually. All details of the products are provided in Supplementary File 1 (full NMR spectra, optical purity, and HRMS-MALDI data).
The remaining NMR spectral data of the products shown in Table 1 are included in Supplementary File 1 (5a-c, 5e, and 5g).
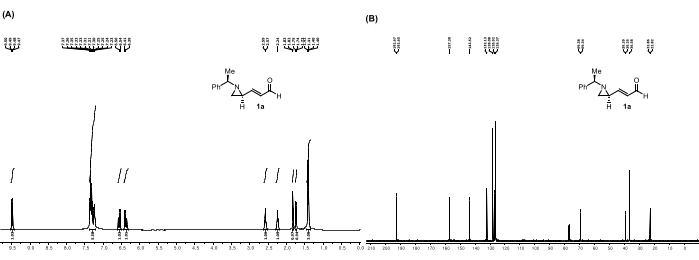
Figure 5: Spectral data for 1a: (A) 1H NMR spectrum. (B) 13C NMR spectrum. Notable peaks in the 1H NMR spectrum: The peaks at 6.56 and 6.38 ppm correspond to the alkene hydrogens in between the aziridine and the aldehyde. Moreover, the peak at 9.47 ppm corresponds to the aldehyde hydrogen. This figure is adapted from Mao et al.28. Please click here to view a larger version of this figure.
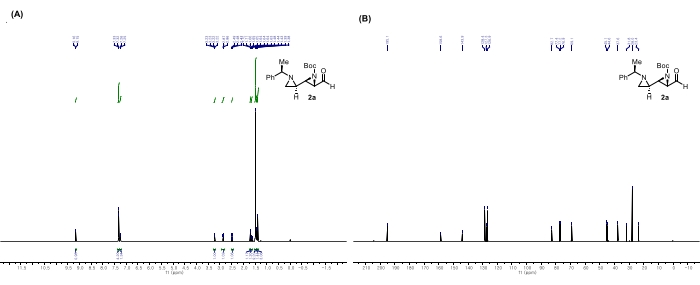
Figure 6: Spectral data for 2a: (A) 1H NMR spectrum. (B) 13C NMR spectrum. Notable peaks in the 1H NMR spectrum: The peak at 9.16 ppm indicates that the aldehyde remains intact. The peak at 1.48 ppm corresponds to the Boc hydrogens. In comparison with the spectrum data of 1a, the peaks of alkene hydrogens have disappeared; however, the peaks of the generated aziridine hydrogens are detected in the range of 1.25-1.72 ppm. This figure is adapted from Rhee et al.52. Please click here to view a larger version of this figure.
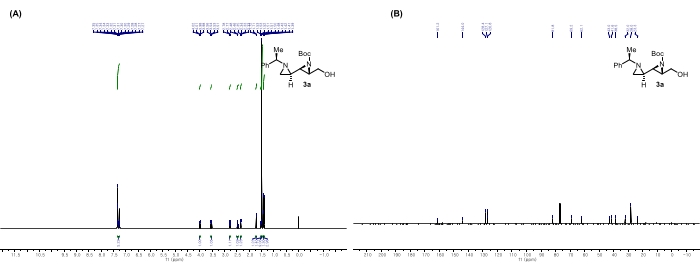
Figure 7: Spectral data for 3a: (A) 1H NMR spectrum. (B) 13C NMR spectrum. Notable peaks in the 1H NMR spectrum: The peak at 1.42 ppm corresponds to the alcohol hydrogen in the ethyl alcohol adjacent to the aziridine, indicating that aldehyde in 2a was reduced to ethyl alcohol. Moreover, the peaks at 4.00 and 3.54 ppm represent the methylene hydrogens in ethyl alcohol. This figure is adapted from Rhee et al.52. Please click here to view a larger version of this figure.
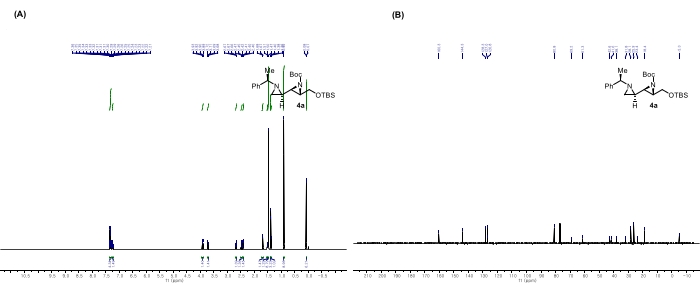
Figure 8: Spectral data for 4a: (A) 1H NMR spectrum. (B) 13C NMR spectrum. Notable peaks in the 1H NMR spectrum: The peaks at 0.90 and 0.07 ppm correspond to the TBS hydrogens. This figure is adapted from Rhee et al.52. Please click here to view a larger version of this figure.
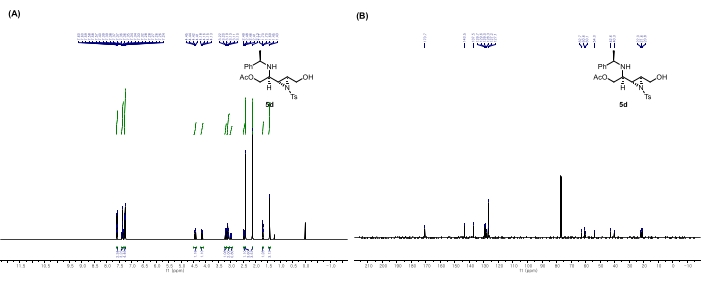
Figure 9: Spectral data for 5d: (A) 1H NMR spectrum. (B) 13C NMR spectrum. Notable peaks in the 1H NMR spectrum: The peak at 2.13 ppm corresponds to the methyl hydrogens of acetate. The peaks at 4.43 and 4.15 ppm correspond to the methylene hydrogens adjacent to acetate, formed after the aziridine ring-opening by acetic acid. Thus, the peaks at 2.13, 3.11, 4.15, and 4.43 ppm are direct evidence of the ring-opening reaction. This figure is adapted from Rhee et al.52. Please click here to view a larger version of this figure.
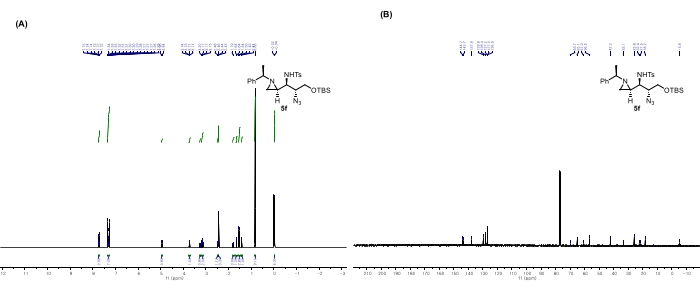
Figure 10: Spectral data for 5f: (A) 1H NMR spectrum. (B) 13C NMR spectrum. Notable peaks in the 1H NMR spectrum: The peak at 4.95 ppm corresponds to the amine proton. The peak at 3.72 ppm corresponds to the hydrogen attached to the carbon bonded to the azide. These peaks are direct evidence of the ring-opening of the aziridine bearing the -Ts group by N3ˉ nucleophile. This figure is adapted from Rhee et al.52. Please click here to view a larger version of this figure.
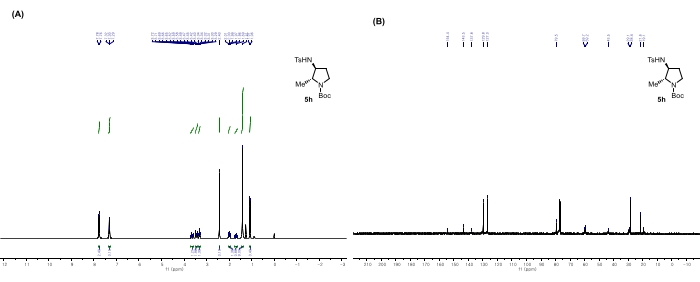
Figure 11: Spectral data for 5h: (A) 1H NMR spectrum. (B) 13C NMR spectrum. Notable peaks in the 1H NMR spectrum: The peaks at 1.70, 1.99, 3.32, 3.45, and 3.65 ppm correspond to the hydrogens of pyrrolidine. The peak of the amine proton adjacent to the -Ts group overlaps with other phenyl groups at 7.30 ppm. These peaks demonstrate the ring-opening and hydrogenation of bisaziridine and the subsequent formation of new cyclic compounds. This figure is adapted from Rhee et al.52. Please click here to view a larger version of this figure.
Supplementary File 1: The structure, NMR spectra, optical purity, and HRMS-MALDI data of the synthetized prodcuts. Please click here to download this File.
