Source: Susannah C. Shissler1, Tonya J. Webb1
1 Department of Microbiology and Immunology, University of Maryland, Baltimore, MD 21201
Immunoprecipitation (IP, also known as a 'pull-down' assay) is a widely used technique that has applications in a variety of fields. First conceived in 1984, it was refined in 1988 (1, 2). The fundamental goal of IP is purification and isolation of a specific protein using an antibody against that protein. The word "immuno" refers to the use of an antibody while the word "precipitation" refers to pulling down a specific substance from a solution. The target protein might be endogenous or recombinant. Most recombinant proteins have an epitope tag (i.e. myc or flag) attached to them to simplify subsequent purification. Typically, it is easier to optimize recombinant protein IP because the antibodies against recombinant epitope tags are very strong and effective. Antibodies against endogenous proteins have extremely variable efficacy – making it much more difficult to optimize these IPs. A necessary step after immunoprecipitation is verification of purification. The isolated protein is resolved using SDS-PAGE and subsequently probed for purity by western blots (Figure 1). An important control is the use of a different antibody during the Western blot to verify pull down of the correct protein. The combination of IP with subsequent techniques is a powerful analysis tool. The goal after purification may be characterization of the protein itself by NMR, mass spectrometry, and in vitro assays, or analysis of the protein's interacting partners (i.e. protein, DNA, RNA) (3, 4, 5).
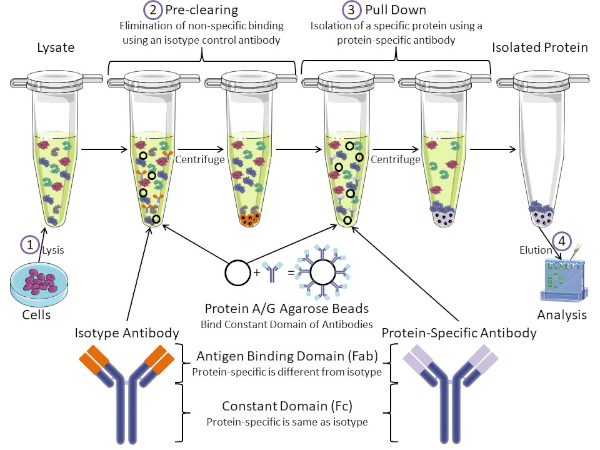
Figure 1: Overview of Immunoprecipitation Procedure. Immunoprecipitation is the isolation of a specific protein using an antibody. After production of lysate from cells, there are two major steps- pre-clearing and pull down. During pre-clearing step, the cell-lysates are pre-cleared of proteins that bind to antibodies non-specifically using an isotype control antibody. In pull down step, the target protein is pulled down using a protein-specific antibody. The isolated protein is then analyzed by Western blot. Isotype antibodies and protein specific antibodies have the same constant domain, but different antigen binding domains. A key component of this protocol is Protein A/G agarose beads that bind the constant domain of antibodies- allowing immunoprecipitation of the target protein. Please click here to view a larger version of this figure.
Antibodies are the key component of an immunoprecipitation that differentiate it from other forms of protein purification (i.e. nickel affinity column purification). Antibodies are molecules made by B cells that can recognize specific protein epitopes. Antibodies have two domains: constant (Fc) and antigen binding (Fab) (Figure 1). The constant domain identifies the type of antibody and dictates function in vivo. Usually, the constant domains of antibodies used for IP are mouse, rat, or rabbit IgG. The antigen binding portion of the antibody recognizes a specific epitope of a specific protein. Antibodies can recognize epitopes on folded proteins that may not exist when the protein is denatured and vice versa. Therefore, the availability of the epitope depends on protein folding – identifying an important factor to consider when choosing antibodies and conditions for IP.
Both prokaryotic and eukaryotic systems have antibody-binding proteins. In eukaryotic systems, the purpose is immune protection from bacteria while in prokaryotic systems, the purpose is protection from the immune system. Antibody-binding proteins affect IP methodology in two ways. First, there is a necessary pre-clearing step (Figure 1) to rid the lysate of proteins that bind antibodies – thereby reducing non-specific binding in the final product. This step uses an isotype antibody that has the same constant domain as but a different antibody binding domain than your protein-specific antibody. Bacterial antibody-binding proteins are the second key component of this method. After the protein-specific antibody binds the target protein, the antibody: protein complex must be pulled down (Figure 1). Proteins A, G, and L are bacterial proteins that bind the constant domain of antibodies. While bacteria use this to subvert the immune system, researchers have co-opted this system for easy antibody purification, and it is used during both the pre-clearing and pull-down steps. These proteins have different binding affinities for different species and different constant domain subtypes – another factor to consider when choosing conditions for IP. Many companies sell Protein A/G labeled agarose beads (Figure 1), pre-made spin columns, or resins to make columns. In general, beads and spin columns are used for smaller sample sizes while resins are used for bulk purification.
In this lab exercise, we demonstrate how to purify the endogenous protein c-myc, from primary murine thymocytes, using Protein A/G Plus agarose beads based basic immunoprecipitation technique. The protocol starts from cell lysate preparation and ends with the with verification of successful protein pull down using Western blot analysis.
The results of the procedure detailed above are shown in Figure 2. From left to right, the lanes contain the control group (isotype), the test group (c-myc), the pre-cleared lysate (lysate), and the molecular weight ladder (ladder). The 25 and 75 kDa ladder bands are marked. The two prominent bands at ~25 kDa and 50 kDa are the light and heavy chain of the binding antibody, respectively and are non-specific to the IP or the samples. c-myc protein which runs around 67kDa on Western blots and is usually visible just below the 75 kDa ladder band. In this blot, the c-myc band is visible in the second lane, but absent in the first lane, indicating that the IP antibody successfully pulled down c-myc. There is no visible band in the pre-cleared lysate lane, suggesting that this protein has low endogenous expression levels.

Figure 2: Results of a Western Blot Analysis, used to assess the purification of c-myc by immunoprecipitation. A band at 67 kDa, corresponding to c-myc, is visible in the anti-c-myc lane, but not the isotype control lane. Note that c-myc levels were not high enough to be visualized in the lysate lane. Please click here to view a larger version of this figure.