Evaluation of SAC responsiveness by nocodazole treatment
The purpose of this experiment is to evaluate SAC activation and strength. By using nocodazole to depolymerize spindle microtubules, all kinetochores will be unattached, which will cause a SAC-mediated cell-cycle arrest. In the present imaging system, DMSO-treated control oocytes extruded a polar body around 14 h after release from milrinone (Figure 1, top panels). Consistent with SAC activation, nocodazole-treated oocytes arrested in metaphase I and did not extrude a polar body (Figure 1, middle panels). To demonstrate that this assay is a reliable indicator of SAC activity, reversine, an MPS1 inhibitor, was used to prevent SAC activation21. When the SAC cannot activate, oocytes extruded a polar body despite having no KT-MT attachments (Figure 1, bottom panels). Oocytes with a weakened SAC will have a mix of outcomes-some oocytes arrest and some extrude polar bodies17. This method of maturing mouse oocytes with and without nocodazole can be used as a first and easy step to challenge SAC activation.
Pattern of Securin-gfp degradation during meiotic maturation
One of the downstream events following SAC satisfaction and silencing is the activation of the APC/C, leading to Securin degradation22. Therefore, evaluation of the pattern of Securin degradation is a direct read-out of SAC function and should be conducted to determine if the nocodazole result was a SAC-dependent perturbation. This assay involves ectopic expression of Securin-gfp into oocytes and subsequent live-cell imaging. After making a curve that follows Securin degradation, three parameters can be determined: (a) The start time of degradation (silencing); (b) the time it takes to reach the minimum Securin level; and (c) the rate of degradation (Figure 2A). In the control, DMSO-treated oocytes, Securin-gfp intensity starts to decrease at ~10 h after milrinone wash out (Figure 2B,C). When oocytes were matured in the presence of the SAC-activating agent nocodazole, Securin-gfp levels were stable during the entire period of meiotic maturation. In contrast, when preventing SAC activation with reversine treatment, the Securin-gfp pattern accelerated; degradation began at ~4 h after milrinone wash-out, consistent with SAC inhibition (Figure 2B,C).
Recruitment of MAD2 to kinetochores by immunofluorescence during meiotic maturation
If the data support a defect in the SAC function, the next step is to evaluate the recruitment of key SAC mediators. The SAC is activated in response to unattached kinetochores. Kinetochores are often unattached in early prometaphase as the spindle is building, and improper attachments are destabilized for correction. Once kinetochores are stably attached to microtubules, the SAC is silenced to allow anaphase onset22. An early step in the SAC response is the recruitment of a series of proteins to kinetochores which serve as a platform for MCC formation22,23. Evaluation of the localization of these SAC components to kinetochores is another strategy to evaluate SAC response. For example, evaluation of MAD2, an MCC component, is a common approach. Confocal microscope images detecting centromeres (ACA) and MAD2 of oocytes matured to early prometaphase I, late prometaphase I, and metaphase I can be used (Figure 3A). To evaluate the strength of the SAC signal, quantify the KT-localized MAD2 pixel intensity (Figure 3B). Significant levels of MAD2 recruitment to KTs occur in early prometaphase I when most KTs are not attached to MTs. The levels of MAD2 reduced in later prometaphase and were nearly absent at metaphase I when all KTs were stably attached to MTs (Figure 3A,B). Therefore, this method can evaluate SAC response when combined with the other two assays.
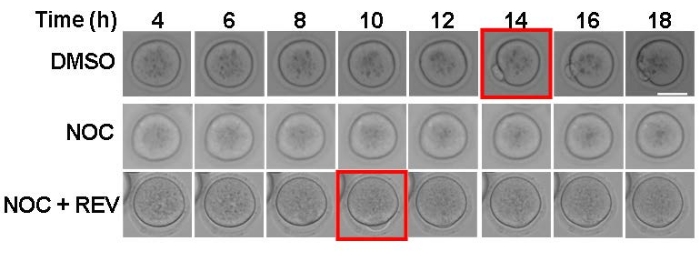
Figure 1: Assessing SAC activation with nocodazole. Representative images from time-lapse imaging of oocytes matured in the presence of DMSO (control), 5 µM nocodazole (NOC), or 5 µM nocodazole + 0.5 µM reversine (NOC + REV). The red square indicates the time frame of polar body extrusion. Two independent experiments were analyzed. Scale bar = 50 µm. Please click here to view a larger version of this figure.
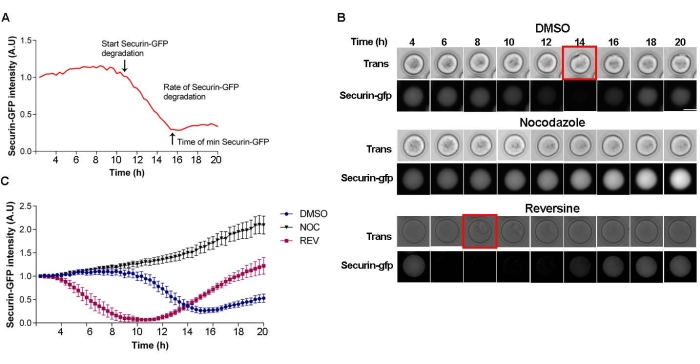
Figure 2: Securin-gfp degradation pattern. (A) Schematic of Securin-gfp degradation curve with three parameters that can be examined. (B) Representative images from a time-lapse of oocytes microinjected with Securin-gfp cRNA and matured in the presence of DMSO (control), 5 µM nocodazole, or 0.5 µM reversine. The red square indicates the time frame of polar body extrusion. Scale bar = 50 µm. (C) Representative Securin-gfp degradation curve of oocytes microinjected with Securin-gfp cRNA and matured in the presence of DMSO (blue circles), 5 µM nocodazole (pink squares), or 0.5 µM reversine (black triangles). Error bars = standard error. # of oocytes per treatment: DMSO = 10; NOC = 15; REV = 5. Please click here to view a larger version of this figure.
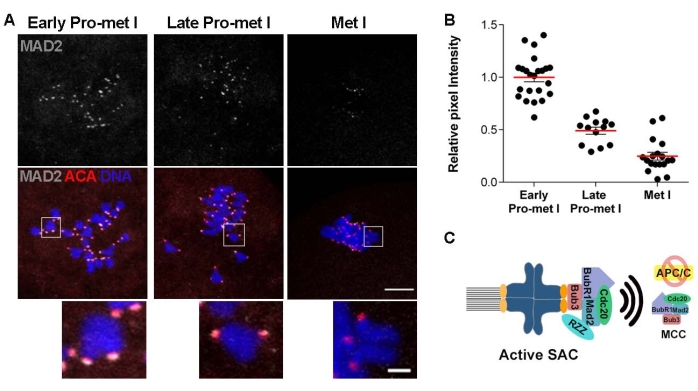
Figure 3: MAD2 recruitment to kinetochores during oocyte meiotic maturation. (A) Representative confocal images of oocytes matured to early prometaphase I, late prometaphase I, and metaphase I, immunostained to detect MAD2 (gray) and kinetochores (ACA) (red). Scale bar = 10 µm; inset: 2 µm. (B) Quantification of the relative intensity of MAD2 from images in (A). Error bars = standard error. (C) Schematic of an unattached kinetochore and SAC activation pathway. # of oocytes per time point: Early prometaphase I = 23; Late prometaphase I = 13; Metaphase I = 18. Please click here to view a larger version of this figure.
