Correlation between polymorphic transition of lipid and API release in lipid-coated API crystals:
API crystals coated with glycerol monostearate are measured via DSC and x-ray directly after coating and after 3 months of storage under accelerated conditions (40 °C, 75% relative humidity)7. Glyceryl monostearate is a multiphasic system containing 40%-55% monoglycerides, 30%-45% diglycerides, and 5%-15% glycerides, mainly tristearin19. The polymorphic forms of sub-α, α, β-prime, and β are reported for monostearin20. Tristearin and 1, 2-distearin show α, β-prime, and β polymorphic forms14.
The DSC data of T0 samples and samples stored under accelerated conditions are shown in Figure 3A. The heating cycle on the T0 sample showed a wide endotherm event up to 10 °C, which can be correlated to the reversible sub-α / α transition described for 1-monostearin and 1-monopalmitin21. Two endothermic events at To = 54.0 °C and 46.7 are correlated to the β-form and a coexisting phase of lower melting point. A coexisting phase can be seen in the x-ray data as the short d-spacing of 4.16 Å corresponding to the polymorphic α-form, organized in a lamellar phase of 57.3 Å and corresponding to different components of the mixture. The lamellar arrangement of lipid coating of T0 sample is given due to the available harmonic peak at 18.7 Å corresponding to the third order reflection in SAXS diffractogram7 (Figure 3B).
DSC data of samples after 3 months of storage under accelerated conditions show an endotherm at To = 55.7 °C, with two overlapped events at Tm = 60.2 °C for the remaining α-form and at Tm = 63.8 °C as the main event for the melting of β-form. The polymorphic transition is confirmed with the x-ray data by detecting short d-spacings of 4.66 Å, 4.58 Å, 4.37 Å, 3.92 Å, and 3.83 Å, typical for the β-form, combined with the reduction in the lamella thickness from 57.3 Å to 50.4 Å, due to the molecular tilting7.
Comparing the release profile of API from coating at T0 and after 3 months of storage under accelerated conditions (Figure 3C) show significant alteration in the release profiles, which can be explained by the evident polymorphic transformation of α-form to β-form with a denser sub-cell arrangement, resulting in a water repellent surface7,21.
Correlation between crystal growth of lipid coating, potential phase separation, and release alteration in lipid-coated API crystals: API crystals are coated with a mixture of tripalmitin and polysorbate 65 in 90:10 %w/w ratio. Tripalmitin is a triglyceride with a purity of 99%5. Triglycerides commonly show the α, β-prime, and β polymorphic forms, ordered by the increased density of crystal pack and increased stability.
Polysorbate 65 is an emulsifier with a hydrophilic-lipophilic balance (HLB) value of 10.5 and a melting temperature of 32 °C. Triglycerides are commonly crystallized in their α-polymorph from the melt. Certain additives induce the transformation of α to β of TAGs, among them polysorbate 65. Moreover, polysorbate 65 acts as an impurity in the system, causing heterogeneous crystallization of tripalmitin at lower driving forces and triggering crystal growth.
The DSC and x-ray data of T0 samples and samples stored under accelerated conditions are shown in Figure 4A,B. The heating cycle of DSC measurements on the T0 sample shows a sharp endothermic event with a peak at 64.8 °C, corresponding to the polymophic β-form of tripalmitin5. This is also detectable in the WAXS region, showing the short-spacing at 4.6 Å, characteristic of the sub-cell of the β-form (Figure 4A,B). The data show, clearly, the induced polymorphic β-form of tripalmitin in the presence of polysorbate 65 at T0 samples and, of course, in stored samples. The corresponding lamellar thickness is calculated using Bragg's equation as d = 2π/q001 = 42 Å5.
The crystal thickness (D) of T0 samples and stored samples can be measured using the Scherrer equation described above. The calculations show a crystal thickness of 24 nm in T0 samples and an increased thickness of 37 nm in stored samples, corresponding to 5.7 and 8.8 lamellae, respectively.
Comparing the release profile of API from coating at T0 and after 3 months of storage under accelerated conditions again shows significant alteration in the release profiles after storage (Figure 4C).
Due to the fact that the mixture of tripalmitin and polysorbate 65 is a two-phasic system, the crystallite growth of tripalmitin is triggered by the existence of polysorbate phase, specially under the accelerated condition (40 °C, 75% relative humidity) where polysorbate 65 is in its liquid molten form. The phase transition and the growth of polysorbate phase under accelerated condition are most probably due to motion of liquid material due to the capillarity and gravity forces5,22. The consequence is the alteration in the API release from coating5.
Correlation between stable solid-state of lipids and stable performance of lipid-based pharmaceutical products: Two different lipid-based pharmaceutical products are assessed: (a) a solid dosage form composed of API crystals coated with lipid-based excipients17 and (b) a liquid dosage form composed of suspended solid-lipid nanoparticles loaded with an API18. The LBEs employed for both products are polyglycerol esters of fatty acids (PGFAs), a group of lipid molecules consisting of oligomeric hydroxyethers of glycerol fully or partially esterified with fatty acids. PGFAs are characterized by monophasic crystallization into α-form, absence of polymorphic transitions, and overall stability of their molecular, nano, and microstructure23.
In the first product, API crystals were coated with PG3-C16/C18p, a PGFA composed of 3 glycerol units partially esterified with palmitic and stearic acid. The DSC and x-ray data of T0 and 3-month stored samples under accelerated conditions are shown in Figure 5. DSC analysis (Figure 5A) shows a single melting peak in the first heating cycle corresponding to the existence of only one polymorphic form of PG3-C16/C18p with To = 54.2 °C. The cooling cycle reveals the monophasic crystallization of the lipid through the existence of one single peak with Tc = 45.4°C. Stored samples reveal unchanged thermograms too, which depict no polymorphism and no phase separation. The stable solid-state of PG3-C16/C18p is confirmed by the SWAXS patterns (Figure 5B). WAXS region shows a peak corresponding to a short-spacing of d = 4.15 Å in T0 and stored samples17. Such short d-spacing is associated with the α-form of TAGs1,13. The unaltered WAXS signal after storage confirms the absence of polymorphism of PG3-C16/C18p. SAXS region reveals a main peak at a long d-spacing of d = 63.7 Å, corresponding to a lamellar structure with 2L-configuration. The crystallite size (D) of T0 samples obtained via Scherrer analysis depicts 23 nm, corresponding to four stacked lamellae. No alterations of lamella thickness (63.5 Å) or crystal growth (four lamellae) are shown after storage. A comparison of the release profile of T0-samples and after storage (Figure 5C) shows the developed product's outstanding stability. The stable solid-state of the lipid matrix provided by PG3-C16/C18p results in the stable performance of the release profile of the product17.
For the second product, API-loaded solid lipid-nanoparticles (SLN) in the form of aqueous nanosuspension were prepared using PG2-C18f as lipid matrix and Poloxamer 188 as emulsifier18. PG2-C18f is a PGFA molecule composed of 2 glycerol units fully esterified with stearic acid. Poloxamer 188 is a non-ionic block polymer with a high HLB of 29. The chemical structure is composed of polyoxypropylene and polyoxyethylene parts. The API is encapsulated into the lipid matrix. Within this product, the solid-state of the lipid can be impacted not only by the processing conditions but also by water-nanoparticle interactions and emulsifier-lipid interactions. The DSC and X-ray data of nanosuspensions at T0 and after 3 months of storage under accelerated conditions are shown in Figure 6. DSC analysis shows an endothermic event at To = 55.3 °C followed by a wide endotherm extended up to 100 °C. The first event attributed to the melting of SLN of PG2-C18f and the wide endotherm is due to water evaporation. Since Poloxamer 188 is dissolved in the water phase, no endotherm is depicted in the first cycle. Stable thermal behavior is depicted in the DSC analysis of stored samples, which show no alterations. Although lipid polymorphism is usually accelerated in nanosized systems, SWAXS analysis confirms the stable behavior of the lipid matrix. The measured short d-spacing of 4.15 Å for PG2-C18f after its crystallization in the freshly manufactured SLN and after 6 months storage of samples under accelerated condition (6m/AC) indicates the presence of the stableα-form. The lamella thickness of PG2-C18f within SLN at T0 (56.5 Å) and after storage (56.3 Å) shows no alterations. The lamellar structure of the lipid is evidenced by a harmonic signal at 18.7 Å. The crystallite size (D) of PG2-C18f by Scherrer analysis is found to be 10.8 nm (two lamellae), showing no crystal growth after storage of the nanosuspensions (11.7 nm, two lamellae)18. The use of SLN in the pharmaceutical industry has been hindered due to well-reported stability issues after storage, such as particle agglomeration and gelation, loss of encapsulation (API expulsion), and unstable release profile. Instead, the application of a stable lipid matrix, PG2-C18f, as herein shown, results in the product performance presented in Figure 7. No particle agglomeration, stable release profile, and stable encapsulation efficiency are depicted. The general instability of SLN has been attributed to lipid polymorphism and other solid-state transitions24. Polymorphic lipids suffer transitions during storage from less dense crystal forms (α-form) to more dense (β-prime and β). This polymorphic transition can affect the surface area of manufactured nanoparticles, especially, if the surface area is not sufficiently stabilized by emulsifier. The consequent can be instabilites such as agglomeration or gelation. Also, the alteration of crystal density from α to β causes the loss of sufficient space for the API within the lipid matrix, which leads to API expulsion, alterations in the encapsulation efficiency, and the release profile. Considering the small size of SLN (in this study x50 = 234.3 nm), the effects of crystal growth on product performance become critical too. The use of a lipid matrix with a stable solid-state resulted in stable product performance18.
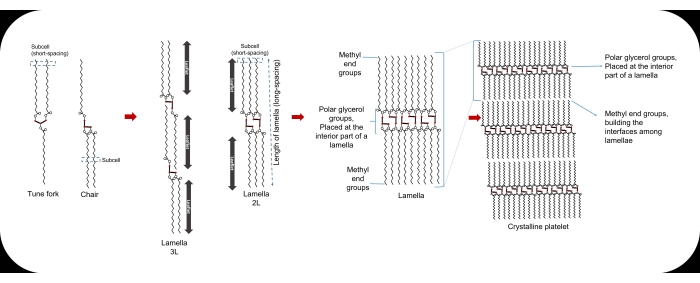
Figure 1: The tune fork and chair configurations of a TAG molecule, the sub-cell, the lamella, and the crystalline platelet. Please click here to view a larger version of this figure.
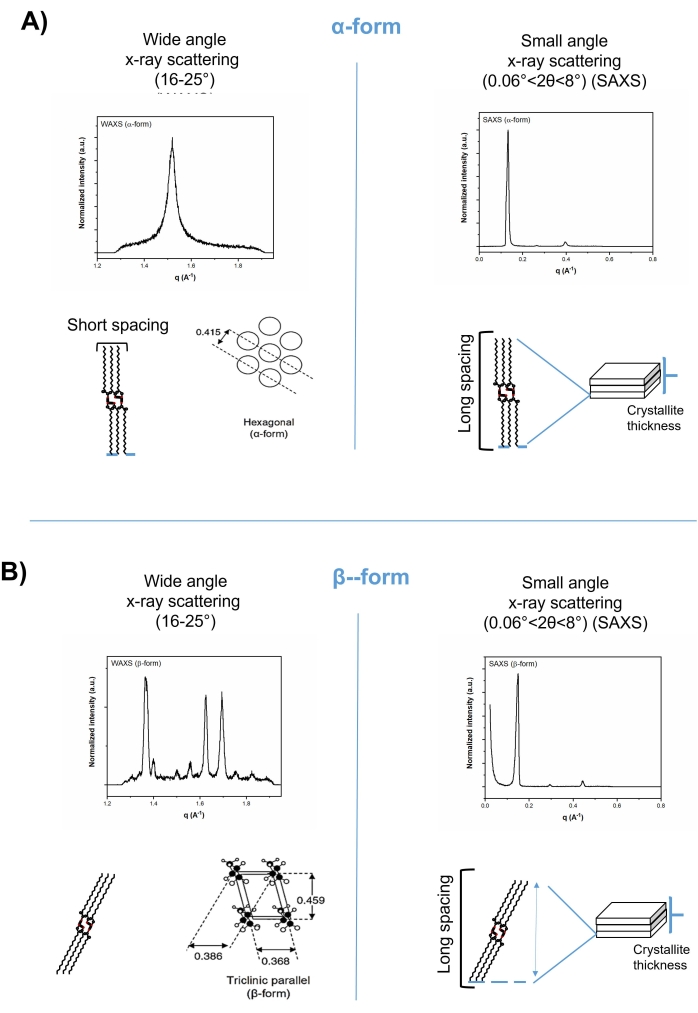
Figure 2: Short spacing (left-hand) and long spacing (right-hand) patterns of tripalmitin in wide-angle and small-angle regions of x-ray diffractograms, respectively. (A) The alpha-form, and (B) the beta-form. Please click here to view a larger version of this figure.
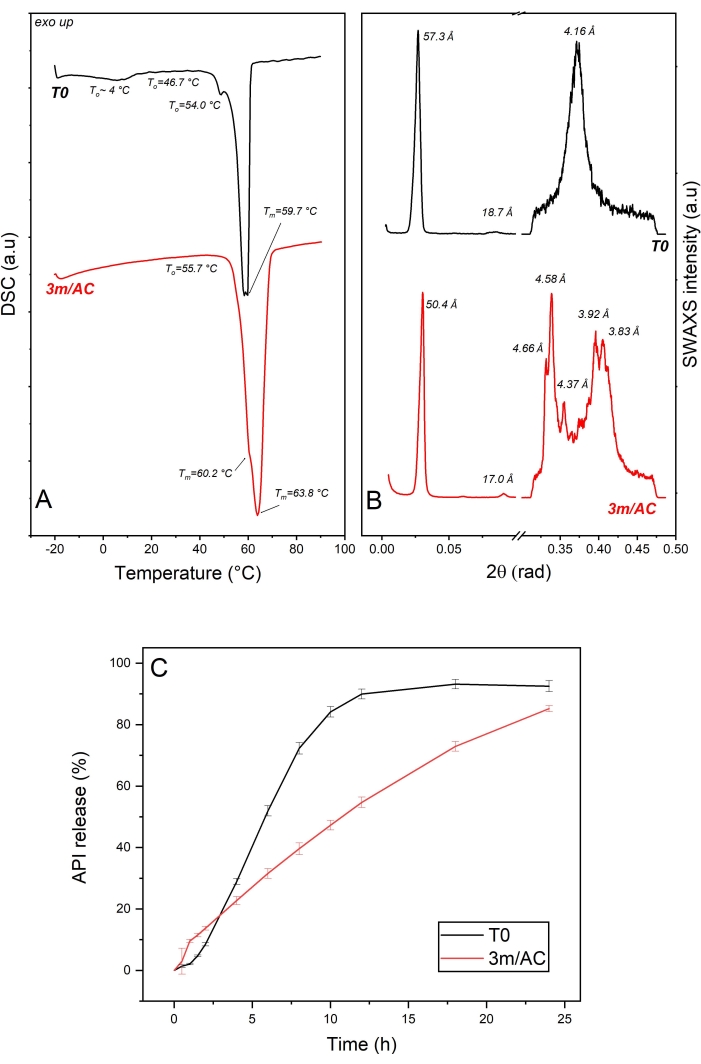
Figure 3: API crystals coated with glycerol monostearate: solid state analysis of lipid as coating material and API release profile of freshly prepared samples (T0) and after 3-month storage under accelerated conditions (AC). (A) DSC, (B) SWAXS, and (C) release profile. This figure has been modified from7. Please click here to view a larger version of this figure.
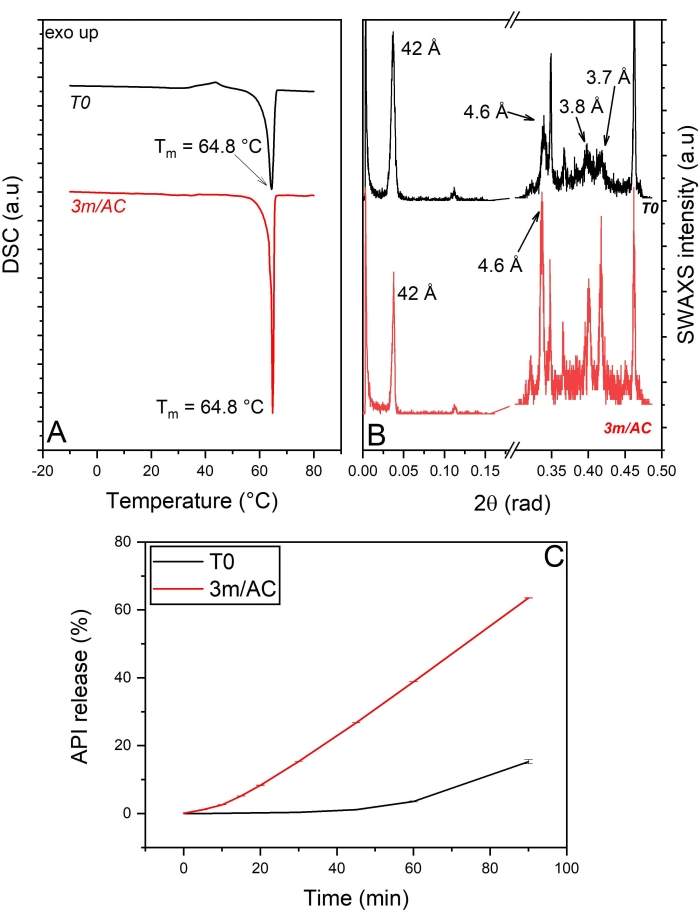
Figure 4: API crystals coated with tripalmitin and polysorbate 65 (90:10 %w/w): Solid state analysis of coating material and API release of freshly prepared samples (T0) and after 3-month storage under accelerated conditions (AC). (A) DSC, (B) SWAXS, (C) API release profile. This figure has been modified from5. Please click here to view a larger version of this figure.
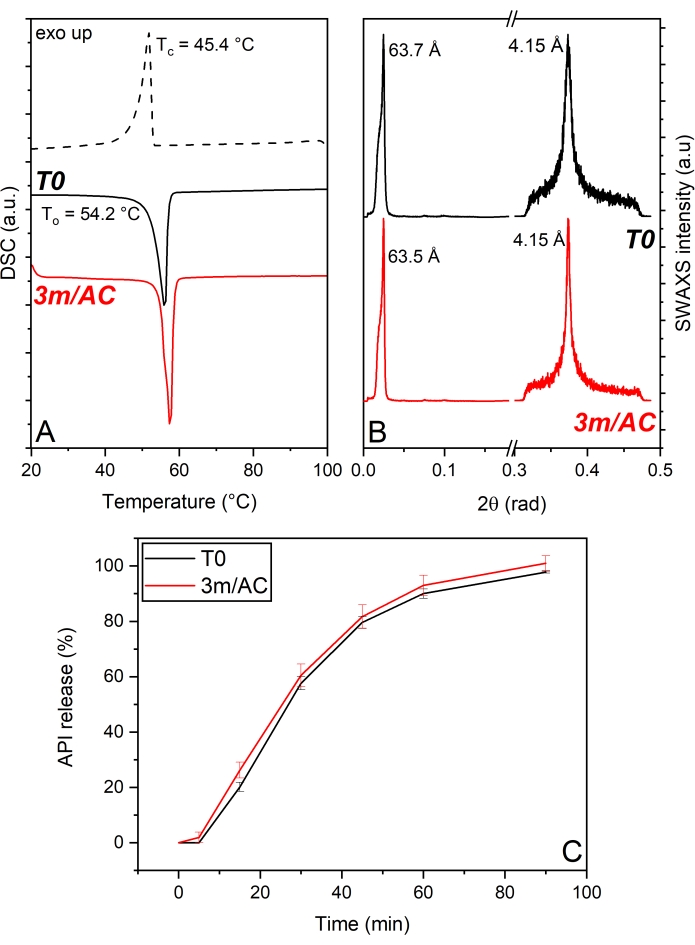
Figure 5: API crystals coated with PG3-C16/C18p: Solid state analysis of PG3-C16/C18p as coating material and API release profile of freshly prepared samples (T0) and after 3-month storage under accelerated conditions (AC). (A) DSC, (B) SWAXS, (C) API release profile. This figure has been modified from17. Please click here to view a larger version of this figure.
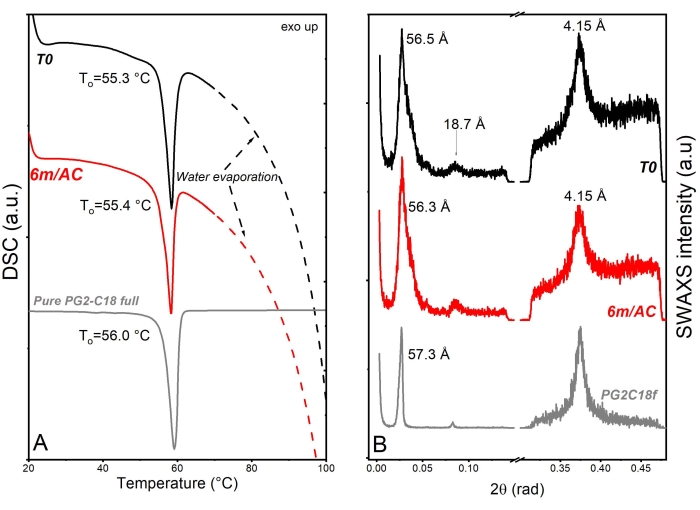
Figure 6: Solid state analysis of freshly prepared SLN samples (T0), after 3-month storage under accelerated conditions (AC), and raw lipid excipient. (A) DSC and (B) SWAXS. This figure has been modified from18. Please click here to view a larger version of this figure.
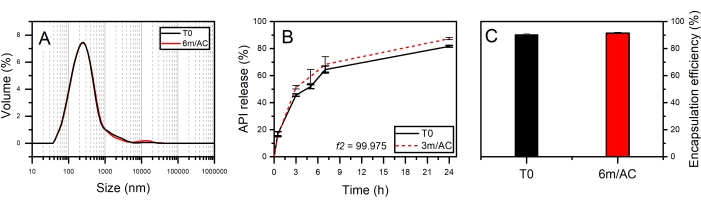
Figure 7: Product performance of freshly prepared SLNs (T0) and after 3 and 6-month storage under accelerated conditions (3m/AC, 6m/AC). (A) Particle size distribution, (B) release profile, (C) encapsulation efficiency. This figure has been modified from18. Please click here to view a larger version of this figure.
