After image acquisition, the representative cell morphology was analyzed using embedded statistics and classifying scripts within the analysis software. The collected data (Figure 6A) reflects that neuron 2 has a larger dendritic structure with a higher density of spines. As a whole, the data suggests that neuron 2 has a more complex dendritic structure compared to neuron 1. To substantiate this result, standard Sholl analysis was performed, which affirms that neuron 2 is more dendritically complex than neuron 1 as denoted by the increased number of Sholl intersections at 50-100 µm from the soma (Figure 6B). Finally, dendritic spines of the two imaged neurons were classified into four main categories based on their overall shapes and sizes. Spines that exhibit more filopodia-like shapes are likely more immature spine subtypes. Spines with defined heads, called mushroom spines, likely contain more developed and mature synapses7. The analysis presented here shows that neuron 1 contains a larger proportion of filopodia-like spines as compared to neuron 2 (Figure 6C). Thus, based on morphology, neuron 2 is more developmentally mature as it is larger, more highly branched, and contains a higher density of mature spines.
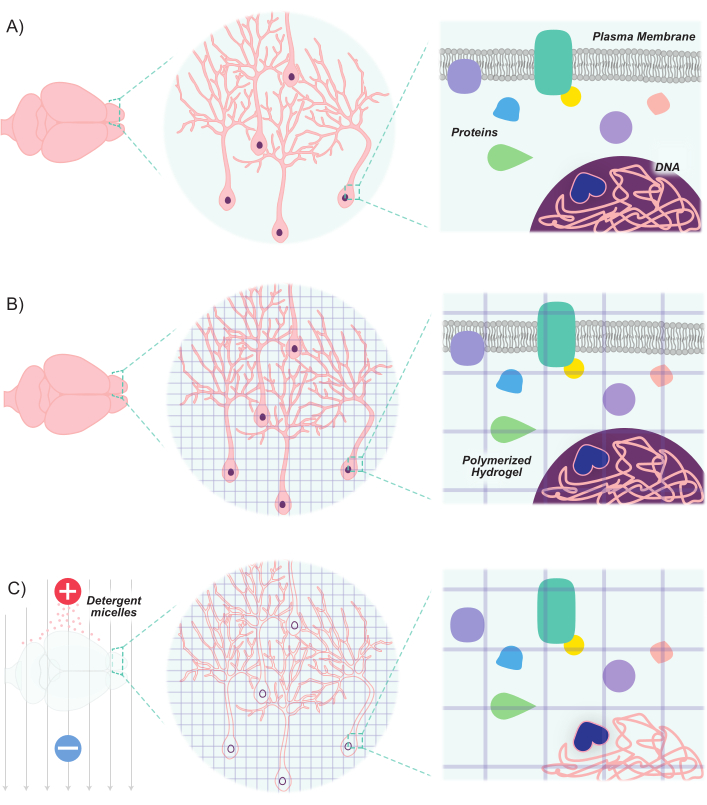
Figure 1: CLARITY protocol renders tissues transparent while maintaining inherent structure and molecule-molecule spatial relationships. (A) Original orientation of neuronal tissue and intercellular components prior to clearing. (B) Hydrogel monomers (purple lines) are infused into the tissue and polymerized into a hydrogel mesh. The tissue and hydrogel mesh are crosslinked via formaldehyde fixation. (C) The tissue is then washed with ionic detergent solutions while exposed to electric fields. During this process, the detergent micelles remove lipid molecules from the tissue, leaving behind a crosslinked network of transparent hydrogel and biomolecules. Please click here to view a larger version of this figure.
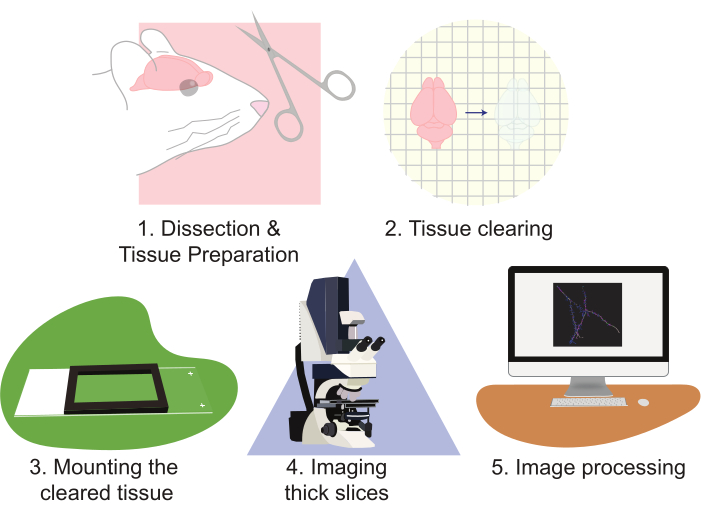
Figure 2: Flow chart of the protocol, diagramming the tissue preparation, clearing, mounting, imaging, and image processing. Please click here to view a larger version of this figure.

Figure 3: Constructing mounting chambers for cleared tissue samples. (A) Cleared whole brain in PBS prior to immersion in refractive index matching solution. (B) Brain slice in refractive index matching solution. (C) Imaging chamber for large and small cleared tissue samples. The chambers can be made using a variety of materials including but not limited to 3D printed plastics and cut conical tubes. (D) Imaging chamber for whole brain or hemisphere imaging, using 50 mL conical tube as a relief before pouring agarose. (E) Whole brain or hemisphere imaging chamber with agarose fully set; this setup is optimal for large barrel immersion objectives. Please click here to view a larger version of this figure.
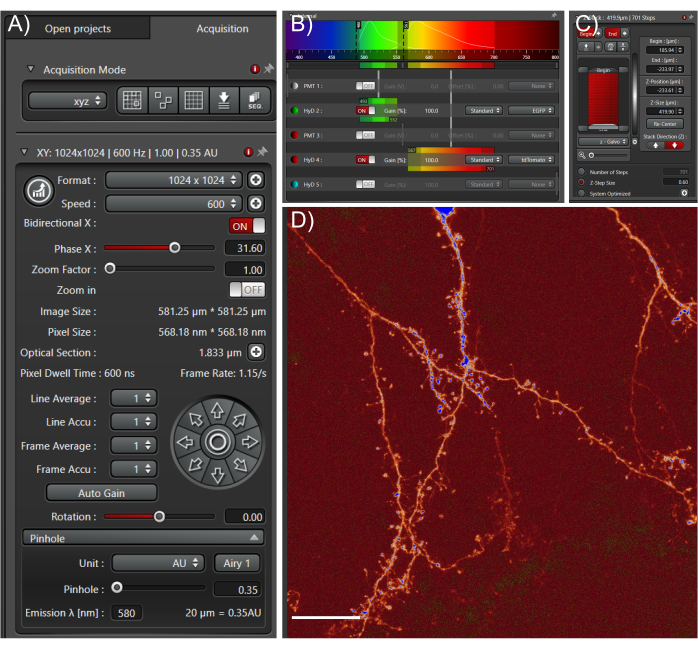
Figure 4: Acquire high quality large datasets from cleared tissue samples. (A) Image acquisition settings used for whole cell high-resolution imaging. The pinhole was fully closed to enable fine optical sections. Scan speed was determined empirically based on optimal pixel dwell times. (B) Light path settings: these will depend on the fluorophore and equipment used. (C) Z-stack settings; a fine z-step was used to capture as much information in the z-direction as possible. (D) Representative max projection; scale bar represents 25 µm. Please click here to view a larger version of this figure.
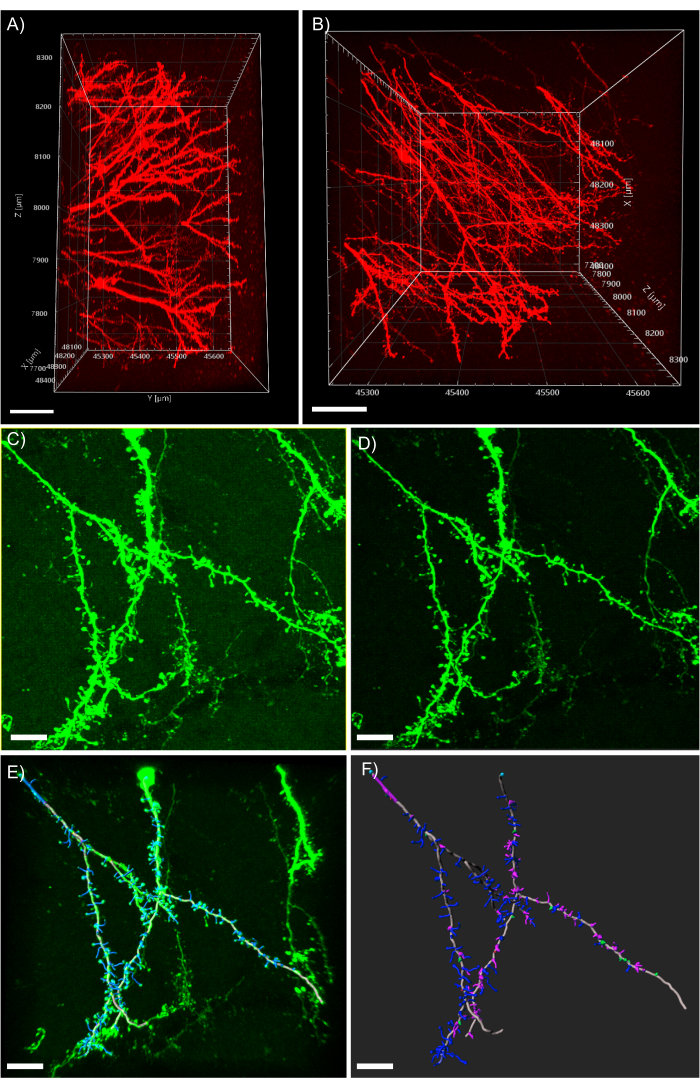
Figure 5: 3D analysis of dendrites in the analysis software. (A) Side-view of a two-photon acquired 600 µm z-stack acquired from a 1 mm thick tissue expressing tdTomato; scale bar represents 100µm. (B) Top-view of the same z-stack acquired in panel A; scale bar represents 100µm. (C) Confocal acquired image of tissue expressing EGFP. Image files can be directly imported into the analysis software and pre-processed for greater image quality; scale bar represents 25 µm. (D) Threshold subtraction is used to remove the consistent background signal present in C. (E) Filament tracing and spine identification: this process is best when performed semi-automatic with the auto-depth feature checked. Spines were then hand labeled after full dendrite reconstruction; scale bar represents 25 µm. (F) Spine classification using a built-in MATLAB extension. Spines have been color coded based on their morphology; stubby spines are red; mushroom spines are green; long thin spines are blue; filopodia are purple; scale bar represents 25 µm. Please click here to view a larger version of this figure.

Figure 6: Representative results. (A) Table of common morphological measurements automatically generated by the selected analysis program. (B) Number of Sholl intersections generated by program statistics. (C) Pie chart representing the distribution of dendritic spine morphologies. Please click here to view a larger version of this figure.
| Solution | Composition | Notes |
| Refractive index matching solution | 80 g of histodenz | pH to 7.5 with NaOH |
| 60 mL of 0.02 M phosphate buffer | Store at 4 °C | |
| 0.01% of sodium azide | Adapted from Marx, V. Nature Methods volume 11, pages 1209–1214 (2014) | |
| Hydrogel | 13.33 mL of 30% Acrylamide (no-bis) | Mix together on ice, otherwise the solution may begin to polymerize |
| 10 mL of 10x PBS | Aliquot and store at -20 °C | |
| 250 mg of VA-044 | ||
| 76.66 mL of ddH2O |
Table 1: Recipes for refractive image matching solution and hydrogel solution. The composition of the refractive index matching solution and the hydrogel are listed.
