Quantitative image analysis
In addition to “just” visualizing cellular processes, live-cell imaging allows to extract quantitative information from the recorded data. Generally, quantitative image analysis is a complex topic whose proper discussion is far beyond the scope of this article, hence, the reader is referred to dedicated textbooks and articles39,40,41. Nevertheless, some basic guidelines associated to the following example data are provided. Several crucial prerequisites must be met to allow image quantification, including: (1) defined molarities of fluorescent dyes must be applied to all samples to allow accurate relative comparison; (2) image acquisition settings must be adjusted in a way that the emission light detectors are never saturated, otherwise maximum intensities are cut off; (3) image acquisition settings must remain fixed during the course of a coherent experimental set, otherwise artificial intensity changes are introduced; (4) image data must be saved in an information-lossless file format along with the meta information containing all instrument settings; and (5) image analysis should be limited to the minimal number of post processing steps required to extract the desired quantitative information.
Usually, defined standards that would allow absolute quantification of recorded signals are not available in the living cell. Thus, in its simplest form, quantitative image analysis relies on the relative comparison of pixel intensities within the same image or between different images recorded with identical settings. The manufacturer’s microscope control software normally includes basic tools for image post-processing and quantitative analysis, or can be upgraded with additional functions for image segmentation, thresholding, ratio imaging, etc. Several open source image processing platforms, differently suitable for various types of imaging data, are available, including ImageJ (https://imagej.net; https://imagej.nih.gov/ij/), icy (http://icy.bioimageanalysis.org/), CMEIAS Bioimage Informatics (http://cme.msu.edu/cmeias/) and Wimasis (https://www.wimasis.com/en/).
The presented example data was processed and analyzed using the ImageJ platform. Briefly, specific regions in the cell, such as the hyphal tip apex or septa, are marked with sizable area selection tools, and the intensity of all contained pixels is readout with the software implemented “measuring tool”. The intensity data from controls and experimental samples is transferred into a spreadsheet file, mathematically analyzed and prepared as graph. Further details may be found in the cited original publications.
Example data 1: FM 4-64 uptake assays
Fungal samples were cultivated as colonies (step 1.2) and mounted by the inverted agar block method (step 3.1). The final concentration of FM 4-64 was 1.67 µM. Imaging settings: HCX PL APO 63x/1.3 NA glycerol immersion objective on an inverted confocal laser scanning microscope (see the Table of Materials); FM 4-64 excitation at 488 nm and emission at 600–700 nm; one frame every minute for up to 150 min. FM 4-64 uptake assays identified defects in the spatio-temporal organization of endocytosis in gene deletion and gene overexpressing mutants of the fungal-specific Sur7-family protein 2 (Sfp2) of T. atroviride9 (Figure 5).
Example data 2: FM 4-64 co-staining of fluorescent fusion proteins targeted to endocytic compartments
Fungal samples were cultivated as colonies (step 1.2) and mounted by the inverted agar block method (step 3.1). The final concentration of FM 4-64 was 2 µM. Imaging settings: CFI Plan Apo VC 60x/1.2 NA XC water immersion objective on an inverted confocal laser scanning microscope (see the Table of Materials); GFP excitation at 488 nm and emission at 500–530 nm, FM 4-64 excitation at 488 nm and emission at 600–700 nm, and bright-field with transmitted light detector, all simultaneously; one frame every 15 s for up to 15 min. FM4-64 co-staining was employed to relate the subcellular distribution of the two enhanced green fluorescent protein (EGFP)-tagged transmembrane proteins Sfp2 and Gpr1 to the endocytic pathway in T. atroviride (Figure 6, Movie 3, Movie 4).
Example data 3: FM 4-64 co-staining for the identification of morphogenetic differences
Fungal samples were cultivated as colonies (step 1.2) and mounted by the inverted agar block method (step 3.1). The final concentration of FM 4-64 was 2 µM. Imaging settings: Plan Apochromat 63x/1.4 NA oil immersion objective on an inverted confocal laser scanning microscope (see the Table of Materials); GFP excitation at 488 nm and emission at 505–550 nm, FM 4-62 excitation at 488 nm and emission at 574–691 nm, and bright-field with transmitted light detector, all simultaneously; one frame every 8.5 s for up to 15 min. FM4-64 co-staining allowed to relate the subcellular localization dynamics of fluorescently labeled BUD-6 polarisome complex protein to endosome trafficking-dependent processes, such as the formation of septa and polarized hyphal tip growth, and characterized differences in the subcellular organization and hyphal architecture between wild type and mutant strains of N. crassa (Figure 7, Movie 5).
Example data 4: Cell wall staining reveals morphogenetic differences
Fungal samples were cultivated as colonies (step 1.2) and mounted by the inverted agar block method (step 3.1). Final concentrations of 2 µM CFW, 20 µM SPF and 100 µM CR were used. Imaging settings: CFI Plan Apo VC 60x/1.2 NA XC water immersion objective on an inverted confocal laser scanning microscope (see the Table of Materials); CFW and SPF excitation at 405 nm and emission at 430–470 nm, CR excitation at 543 nm and emission at 580–620 nm. The different interaction properties of CFW, SPF and CR with cell wall polymers highlight morphogenetic differences between the Δsfp2 mutant and the wild type strain of T. atroviride9. Increasing cell wall stress inflicted by elevated dye concentrations occurs quicker and more pronounced in the mutant compared to the wild type. In addition, the same images allow to quantify morphogenetic differences regarding hyphal diameter and septal distance between both strains (Figure 8).
Example data 5: Real-time monitoring of cell wall biosynthesis
Germlings were cultivated as liquid culture (step 1.4) in 8-well chambered micro-slides (step 3.2). The final CFW concentration was 0.12 µM. Imaging settings: CFI Plan Apo VC 60x/1.2 NA XC water immersion objective on an inverted confocal laser scanning microscope; CFW excitation at 405 nm and emission at 420–470 nm; one frame every 20 s for up to 35 min. The very low CFW concentration prevents saturation of the cell wall with dye molecules and allows quantitative real-time monitoring of cell wall biosynthesis. This reveals that the deposition of new cell wall material is not uniform but responds very rapidly to localized physical stresses resulting from the relative displacement of one cell upon cell-cell attachment prior to germling fusion in N. crassa (Figure 9, Movie 6).

Figure 1: Biochemical and biophysical properties of FM dyes. (A) Chemical structures of FM 1-43/SynaptoGreen C4 and FM 4-64/SynaptoRed C2. (B) Absorption and emission spectra of both FM dyes, overlaid with the optimal imaging settings for membrane-bound dyes in filamentous fungi: 445–495 nm blue light will excite FM 1-43 with 100–80% efficiency, whereas 488 nm of an Argon laser will excite the dye with 91% efficiency. Due to the blue-shift upon membrane binding (*), the optimal detection range of FM 1-43 emission is between 520–590 nm. Similarly, the optimal imaging settings for FM 4-64 in fungi are 471–541 nm (100–80% efficiency) when using a polychromatic excitation light source or 514 nm (99% efficiency) when using an Argon laser, and 650–750 nm for emission light detection. Please click here to view a larger version of this figure.
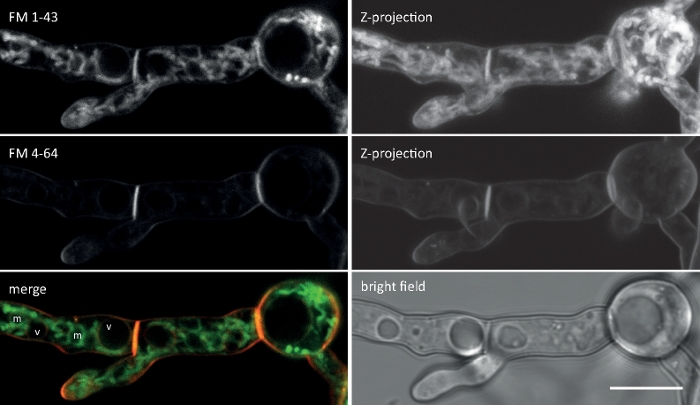
Figure 2: Simultaneous co-imaging of FM 1-43 and FM 4-64. An equimolar mixture of both dyes was added to a liquid germling culture of N. crassa yielding a final concentration of 10 µM. At 25 min after dye addition, FM 1-43 has stained the plasma membrane and already accumulated in internal membranes, including strongly stained mitochondria (m) but largely excluding vacuolar membranes (v), and is more than eight-times stronger compared to FM 4-64 (average fluorescence intensities 176 to 21, respectively), whose lower hydrophobicity/higher hydrophilicity slows down its internalization rate leading to a prolonged dwelling time at the plasma membrane. Scale bar = 10 µm. Please click here to view a larger version of this figure.
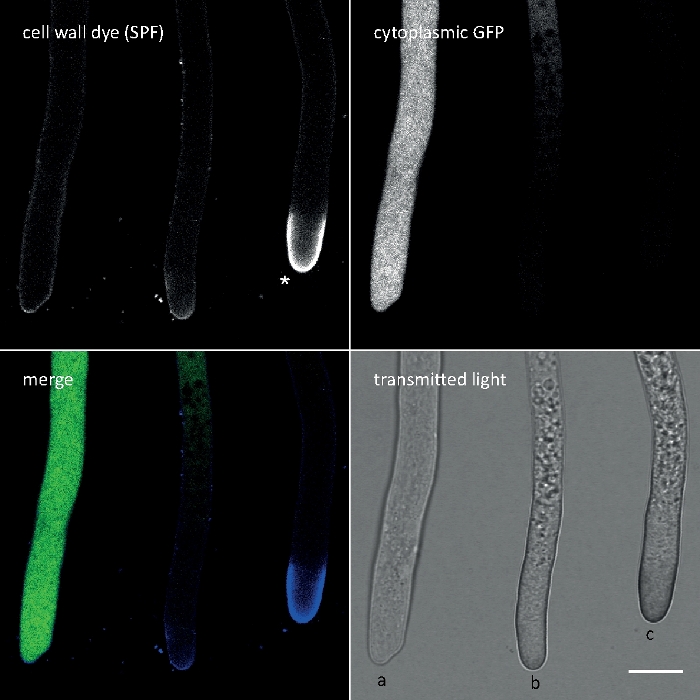
Figure 3: Cell wall stress-induced deposition of cell wall polymers at the tip apex often entails cell autolysis. Hyphae of T. atroviride expressing cytoplasmic GFP were stained with 10 µM Solophenyl Flavine 7GFE 500 (SPF) and imaged immediately upon mounting. Non-stressed hyphae (a), stressed hyphae with slightly increased glucan/chitin deposition at the apex and progressing autolysis (b), and heavily stressed hyphae with pronounced apical glucan/chitin cap (asterisk) and terminal autolysis (c) evident by the total loss of GFP fluorescence and extensive vacuolization. Scale bar = 10 µm. See Movie 1 for full time course sequence. Note that the three hyphae were indeed located next to each other. Please click here to view a larger version of this figure.
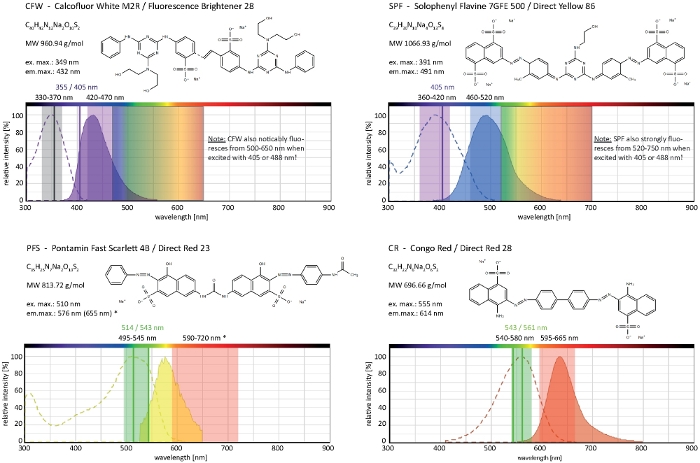
Figure 4: Biochemical and biophysical properties of cell wall selective dyes. Chemical features given are that of the sodium salts of each dye. Absorption and emission spectra correspond to those in cellular environments. Indicated monochromatic laser excitation lines (written in color), polychromatic excitation ranges applicable for epifluorescence microscopes, and emission light detection ranges are those recommended for imaging in filamentous fungi. Two laser excitation lines are indicated when both work equally well. (*) The emission spectrum of cell wall-bound PFS is significantly more red-shifted than previously noted33, however, resulting in very good S/N-ratios with lower dye concentrations than used before. The complete spectrum of CR is currently unavailable, hence that of Nile Red (CAS No: 7385-67-3) is shown as the closest match. Detailed information on the spectral properties of CR can be found elsewhere42. Please click here to view a larger version of this figure.
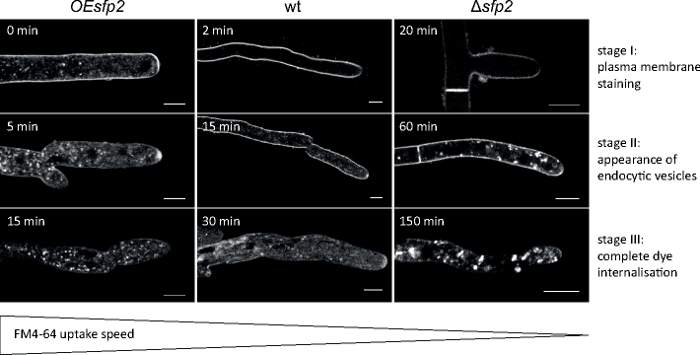
Figure 5: Influence of Sfp2 on the endocytic uptake of FM4-64. Three successive key stages of FM dye uptake are easily discernible in the wild type (wt). Stage I: exclusive plasma membrane staining, stage II: first appearance of FM dye in endocytic vesicles, and stage III: exclusive staining of endocytic vesicles and endomembranes. Equivalent staining patterns are shown at the earliest time point of their appearance. In comparison to the T. atroviride wild type, endocytosis is slightly accelerated in the sfp2 over-expressing mutant (OEsfp2), whereas dye uptake is dramatically delayed in the sfp2 deletion mutant (Δsfp2). For instance, dye uptake into the plasma membrane occurs instantly in OEsfp2 but takes 2 min in the wild type; and complete internalization of FM dye from the plasma membrane occurs 10x faster in OEsfp2 compared to Δsfp2. Scale bars = 5 μm. Figure is reproduced from Atanasova et al.9 in agreement with the Creative Commons License (https://creativecommons.org/licenses/by/4.0/). Please click here to view a larger version of this figure.
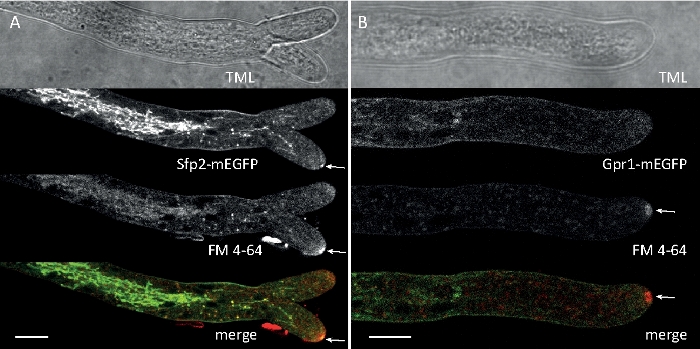
Figure 6: Co-staining of EGFP-labeled membrane proteins with FM 4-64 facilitates differentiation of distinct subcellular localization dynamics in T. atroviride. (A) The four-transmembrane domain protein Sfp2 co-localizes with FM4-64 labeled organelles, including the plasma membrane and septa, the Spitzenkörper (Spk; arrow) and presumable tubular vacuoles. (B) The GPCR-like seven-transmembrane protein Gpr1 co-localizes with FM4-64 to the same organelles as Sfp2, except the Spk. Scale bars, 10 µm. See Movie 3 and Movie 4 for full time course sequences. The figure has been modified from Atanasova et al.9 in agreement with the Creative Commons License. Please click here to view a larger version of this figure.
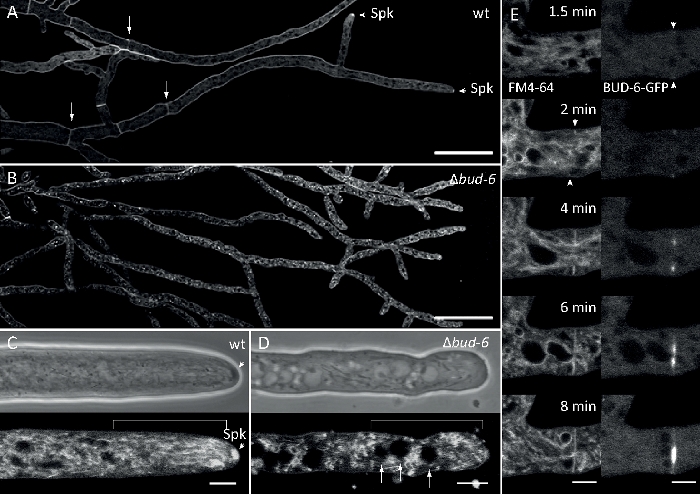
Figure 7: FM 4-64 staining differentiates Δbud-6 mutant from wild type, and localizes BUD-6 at the septal ring. (A) FM 4-64 staining of hyphae of the N. crassa wild type (arrows indicate septa; arrowheads indicate the Spk). (B) Septa and Spk are absent in ∆bud-6. Scale bars, 50 µm. (C and D) Close-up of the hyphal apex and subapex of wild type (C) and ∆bud-6 (D). Spk (arrowhead) differentiates in the wild type but not ∆bud-6. Brackets indicate the nuclear exclusion zone not established in ∆bud-6. Scale bars = 5 µm. (E) BUD-6-GFP recruitment to the incipient septation site preceding plasma membrane invagination (arrowheads) and accompanying septal constriction. Scale bars = 5 µm. See Movie 5a and Movie 5b for complete time course sequences. Figure has been reproduced with modifications from Lichius et al.16 in agreement with the Creative Commons License. Please click here to view a larger version of this figure.
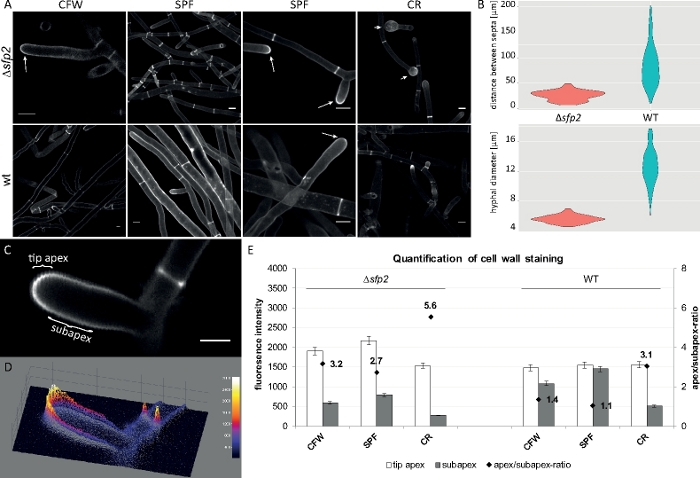
Figure 8: Deletion of sfp2 changes the deposition pattern of cell wall material and affects hyphal morphogenesis of T. atroviride. (A) CFW and SPF staining reveals increased cell wall deposition in Δsfp2 (arrows) compared to the wild type (wt). CR staining induces extensive tip swelling only in Δsfp2 (arrowheads). Scale bars = 10 µm. (B) Morphogenetic defects in Δsfp2 include significantly reduced septal distances (Δsfp2 = 26.0 µm, wild type = 85 µm; n = 60; ANOVA Pr < 2−16) and smaller hyphal diameters (∆sfp2 = 5.6 µm, wild type = 12.6 µm; n = 100; ANOVA Pr < 2−16). (C) Increased dye fluorescence in the tip apex compared to the subapex. Scale bar = 5 µm. (D) Intensity-coded 3D surface plot of (C). (E) Quantification of relative fluorescence intensities in Δsfp2 and wild type (n = 55). Figure has been reproduced from Atanasova et al.9 in agreement with the Creative Commons License. Please click here to view a larger version of this figure.
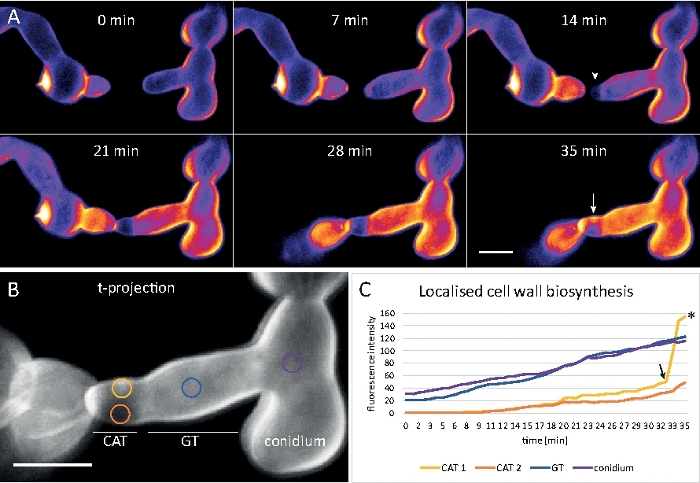
Figure 9: Real time monitoring of cell wall biosynthesis. (A) Conidial anastomosis tube (CAT) fusion between germlings of N. crassa. Physical contact becomes apparent by the germling torque response (21–28 min). Intensity color-coded CFW fluorescence indicates regions with little (dark blue) and intense (yellow) cell wall deposition. The initially unstained homing tip (arrowhead), deposits new cell wall material upon tip contact and in the area experiencing the greatest physical stress (arrow). Scale bar = 5 μm. See Movie 6 for full time course sequence. Figure reproduced from38 with permission. (B) Projection of (A) indicating four circular regions in which fluorescence intensities were measured. Scale bar = 5 μm. (C) Plot of the indicated regions showing the rapid increase in localized cell wall biosynthesis in response to physical stress (CAT 1, arrow). In the germ tube (GT) and the spore body (conidium), cell wall biosynthesis increases steadily. Please click here to view a larger version of this figure.

Movie 1: Dye-induced cell wall stress. 10 µM SPF (cyan) were added to T. atroviride hyphae expressing cytoplasmic GFP (magenta). Extensive tip staining occurs immediately, followed by the rapid lysis of hyphal compartments within 2 min; evident by the disappearance of GFP fluorescence. Please click here to download this Movie.
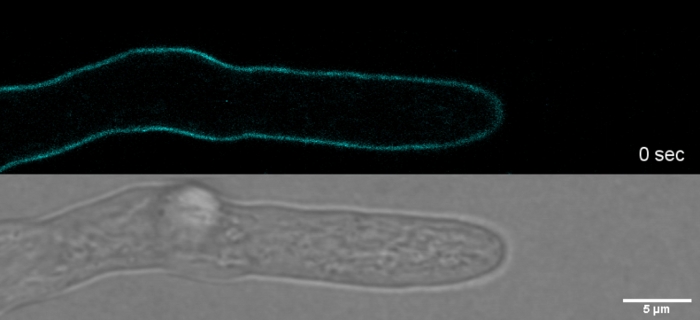
Movie 2: Vital SPF staining. 2 µM SPF (cyan) allow to track tip growth of T. atroviride hyphae with high spatial and temporal resolution without inducing cell wall stress artefacts at the tip apex. Please click here to download this Movie.
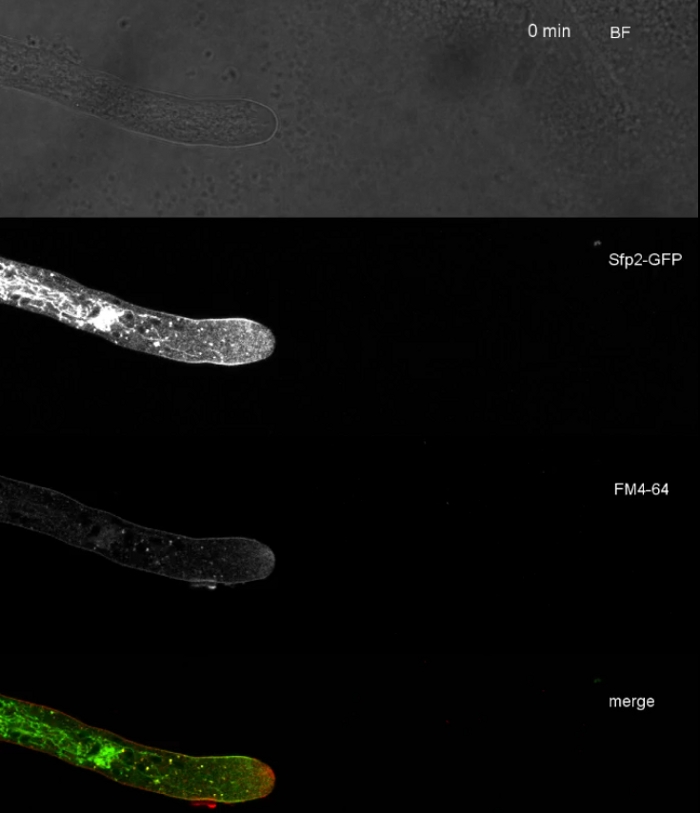
Movie 3: FM 4-64 co-staining of Sfp2-GFP. Co-staining of T. atroviride expressing Sfp2-mEGFP (green) with 1.67 µM FM 4-64 (red) reveal the overlapping and distinct localization of the membrane protein with the endocytic pathway. Please click here to download this Movie.

Movie 4: FM 4-64 co-staining of Gpr1-GFP. Co-staining of T. atroviride expressing Gpr1-mEGFP (green) with 1.67 µM FM 4-64 (red) reveal the overlapping and distinct localization of the membrane protein with the endocytic pathway. Please click here to download this Movie.
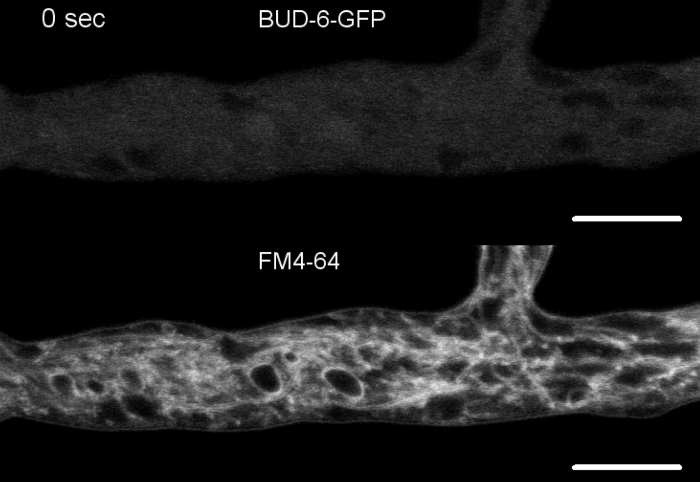
Movie 5: FM 4-64 co-staining of BUD-6-GFP. (5a) Co-staining of N. crassa expressing BUD-6-GFP (green) with 2 µM FM 4-64 (red) allow tracking of BUD-6 dynamics during septum formation relative to the associated plasma membrane invagination. (5b) Cropped and merge image of (5a). Please click here to download Movie 5a
Please click here to download Movie 5b.
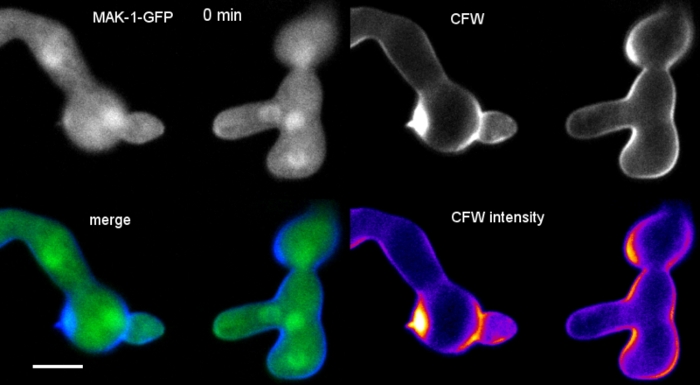
Movie 6: Real-time monitoring of cell wall biosynthesis. N. crassa germlings expressing MAK-1-GFP (green) were co-stained with 0.12 µM CFW (blue) to reveal localized cell wall biogenesis during CAT-mediated germling fusion. Note that there is some bleed through of the CFW signal into the GFP channels, illustrating that SPF or CR are better choices as sequential and simultaneous co-imaging dyes for GFP, respectively. Please click here to download this Movie.
Table 1: Properties of membrane and cell wall selective fluorescent dyes. * = mg/mL values corrected for the reduced purity/dye content to result equimolarity in all solutions; n.i.a. = no information available. Please click here to download this table.
