Here we described the design, synthesis and biological evaluation of a homodimeric pomalidomide-based PROTAC for the degradation of CRBN. Our PROTAC interacts simultaneously with two CRBN molecules and forms ternary complexes that induces self-ubiquitination and proteasomal degradation of CRBN with only minimal remaining effects on pomalidomide-induced neo-substrates IKZF1 or IKZF3.
Out of a series of previously published pomalidomide-based PROTAC molecules24, compound 8 was particularly efficient in the chemical-induced degradation of CRBN. Its synthesis can be accomplished as follows (Figure 4). A 1,1'-carbonyldiimidazole-promoted condensation of Boc-protected l-glutamine leads to the cyclized imide 1. The N-methylated analog 2 is accessible through alkylation with methyl iodide. Both building blocks (1 and 2) are transformed, after N-deprotection under acidic conditions, to phthalimide derivatives (3 and 4) in the course of a ring-opening/recyclization reaction using 3-fluorophthalic anhydride. Thalidomide analogs, in general, are susceptible to hydrolytic decomposition and should only be used in the next step after sufficient drying. Compound 3 is susceptible to an aromatic nucleophilic substitution with primary aliphatic amines25; this conversion was found to proceed efficiently only when dry solvents are used. The design of a true homodimeric product implies the linker connection of two identical functional substructures and the application of symmetrical linker. The linker which is part of PROTAC 8 represents an N-to-N, polyethylene-based linear chain. The corresponding α,ω-diamine 6 leads to the desired final compound 8 when reacted with building block 3 in molar ration of 1:2 in DMSO at 90 °C. Among other analytical data24, the structure of 8 was verified by NMR spectra (Figure 5A). Compound 9, designed as a suitable negative control, has an only minimal, but critical structural deviations, compared to the active homo-PROTAC 8. It is known that N-methylation within the glutarimide portion abolishes CRBN binding26,27. One pomalidomide portion of the negative control compound 9 bears an N-methyl residue. It can be prepared by a first nucleophilic substitution of 3 with the N-monoprotected linker building block 5, followed by cleavage of the Boc protecting group and a subsequent coupling to intermediate 4. Owing to its asymmetrical structure, some of the corresponding carbons showed distinct 13C NMR signals (Figure 5B).
The homo-PROTAC 8 was observed to be highly potent, leading to an almost complete proteasomal degradation of CRBN. The interpretation of CRBN, IKZF1 and IKZF3 protein levels in multiple myeloma cells were confirmed by western blot analysis (Figure 6, Figure 7, Figure 9B), a semi-quantitative standard method, where the change in protein expression can be detected easily. The antibodies used in this paper are of good quality and the method is an optimized standard procedure in our lab.
In addition, degradation of CRBN by compound 8 did not affect cell viability and conferred resistance to IMiDs (Figure 8, Figure 9A), which is in line with CRISPR/Cas9-mediated knockout of CRBN by sgRNAs24. The luminescence signal in the cell viability assay was based on ATP release, which can be interpreted as dead cell count. This method can be easily performed in a short time with a high number of samples. An alternative method for the measurement of viable/dead cells is an Annexin V/ 7-AAD staining by flow cytometry.
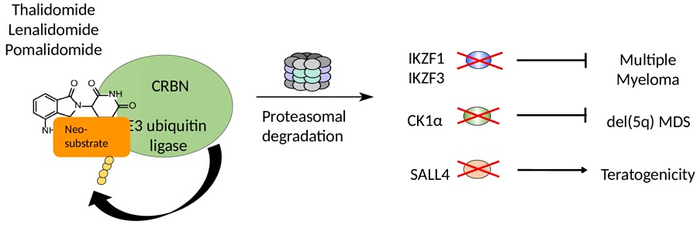
Figure 1: The E3 ubiquitin ligase CRBN is the main target of IMiDs. Immunomodulatory drugs bind to CRBN and recruit several neo-substrates for proteasomal degradation. IMiD-induced degradation of the lymphoid transcription factors IKZF1 and IKZF3 is responsible for the effects on multiple myeloma cells and some of the immunomodulatory properties. Casein kinase 1α is selectively degraded by lenalidomide but not the other IMiDs and contributes to the activity of lenalidomide in myelodysplastic syndrome with loss of chromosome 5q. SALL4 was recently discovered as a common target of all IMiDs that is likely linked to the teratogenicity induced by thalidomide and its analogs. Please click here to view a larger version of this figure.
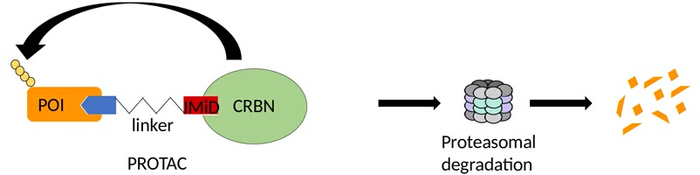
Figure 2: PROTACs degrade the protein of interest (POI). PROTACs are heterobifunctional molecules, where a linker connects a ubiquitin ligase ligand to a POI ligand. By the formation of ternary complexes, a ubiquitin ligase, such as CRBN, then ubiquitinates the POI, resulting in its proteasomal degradation. Please click here to view a larger version of this figure.

Figure 3: Bifunctional homo-PROTAC for the degradation of the E3 ubiquitin ligase CRBN. In a pomalidomide-based homo-PROTAC, two ubiquitin ligase binders are connected to induce cross-ubiquitination of CRBN resulting in a chemically induced knockdown of CRBN. Please click here to view a larger version of this figure.
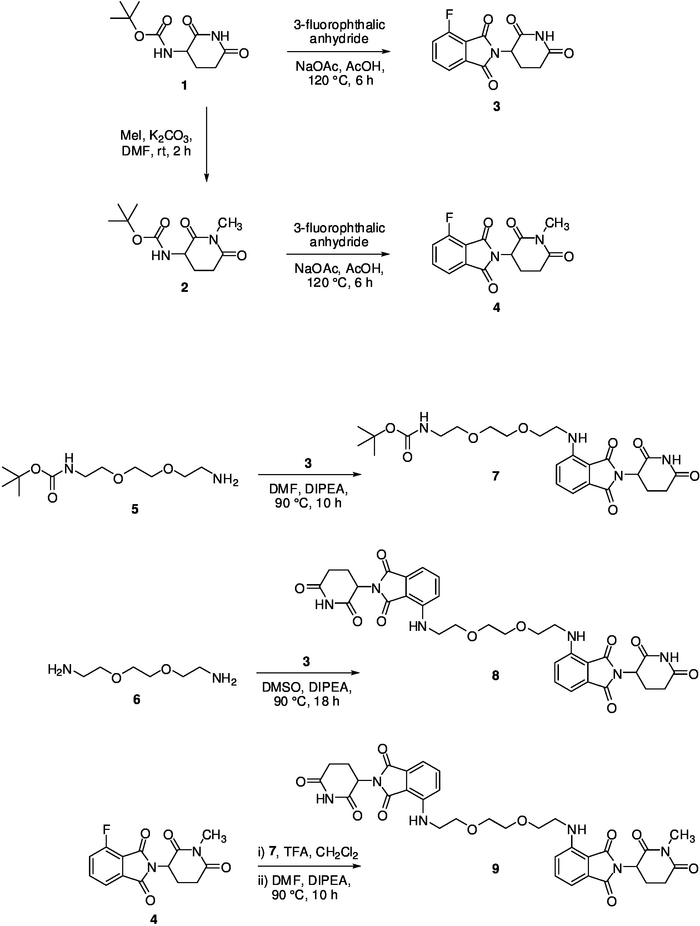
Figure 4: Synthesis of homodimer 8 and heterodimer 9. Please click here to view a larger version of this figure.
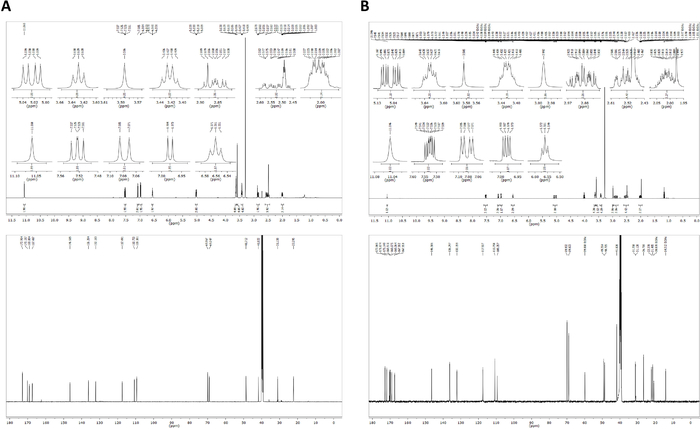
Figure 5: 1H NMR (top) and 13C NMR (bottom) spectra. Spectra of compound 8 (A) and compound 9 (B) were recorded in DMSO-d6 on an NMR spectrometer. Chemical shifts are given in parts per million (ppm). Please click here to view a larger version of this figure.
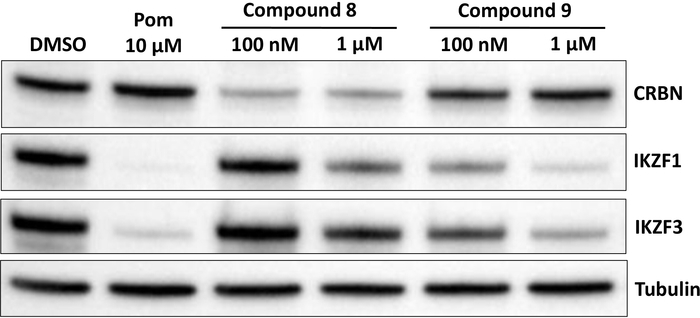
Figure 6: Effects of compounds 8 and 9 on CRBN, IKZF1, and IKZF3. Pomalidomide-based homo-PROTAC compound 8 induces CRBN degradation with weak remaining effects of pomalidomide on IKZF1 and IKZF3. In contrast, compound 9 that contains a methyl group on one of the pomalidomide residues has no effect at the indicated concentrations (µM). MM1S cells were treated for 24 h treatment. Effects on CRBN, IKZF1, IKZF3 and tubulin (loading control) were analyzed by western blot. Please click here to view a larger version of this figure.
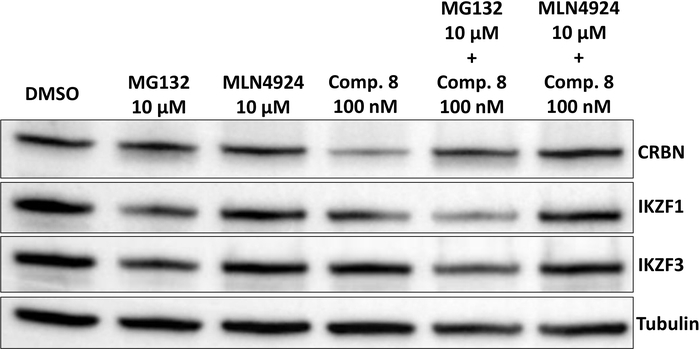
Figure 7: CRBN degradation can be blocked by the proteasome inhibitor MG132 or by MLN4942 that blocks ubiquitin ligases indirectly via neddylation inhibition. The multiple myeloma cell line MM1s was pretreated with 10 µM MG132, 10 µM MLN4924 for 1 h before addition of homo-PROTAC compound 8 at 100 nM for 3 h of combined treatment. Please click here to view a larger version of this figure.
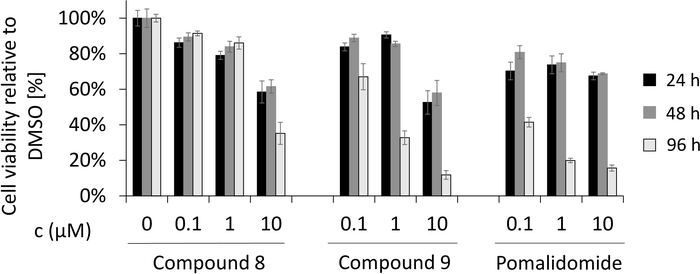
Figure 8: Cell viability assay in MM1S multiple myeloma cells. Effects of compound 8 and negative binding control compound 9 on cell viability in the pomalidomide sensitive myeloma cell line MM1S after 24 h, 48 h and 96 h treatment. Cell viability was measured after 4 days in triplicates. Please click here to view a larger version of this figure.

Figure 9: Compound 8 antagonizes the effect of pomalidomide in multiple myeloma cell lines. Cells were pretreated with 100 nM compound 8 for 3 h. Afterwards 1 µM pomalidomide was added. Cell viability was measured after 4 days in triplicates. *** p <0.001 according to Student´s t-test (A). Western blot analysis for CRBN, IKZF1, IKZF3 and tubulin (loading control) after pretreatment of MM1S cells with 100 nM compound 8 for 3 h, before addition of 1 µM pomalidomide (B). Reprinted (adapted) with permission from Steinebach, C. et al. 201824. Copyright 2019 American Chemical Society. Please click here to view a larger version of this figure.
