To evaluate the efficiency of our protocol and to explore the morphology of the meristems from different species, we have performed the confocal live imaging experiments on the inflorescence meristem from Arabidopsis and the vegetative meristems from both tomato and soybean. In this study, Arabidopsis ecotype Landsberg erecta, tomato cultivar Micro-Tom and soybean cultivar Williams 82 have been used as examples.
Seen from the orthogonal section through the middle of the Arabidopsis SAM, it is clear that PI was able to stain the horizontal walls from almost all the cells at the multiple cell layers (Figure 1A). Shown from one transverse section through the corpus of the inflorescence SAM, the cells from the XY plane are also clearly imaged (Figure 1B). In the 3D projection view, the inflorescence meristem forms a dome like structure and is surrounded by the developing flower primordia, where the cells are also stained and imaged (Figure 1 C-D) (Movie 1).
Seen from the orthogonal section through the middle of the tomato SAM, it is clear that PI was able to stain the horizontal walls from cells at the multiple cell layers, although the PI signal from the deep interior region is slightly lower. From one transverse section through the deep layers of the vegetative SAM, the cells from the XY planes are also clearly imaged, and the boundary formed between the vegetative meristem and leaf primordia is also imaged (Figure 2B). The 3D project view can further provide a comprehensive view of the shape and organization of the vegetative meristem from tomato (Figure 2C-D) (Movie 2).
In the orthogonal view through the middle of the soybean SAM, we can see the dome-like vegetative meristem and its derived new leaf primordia (Figure 3A). In the 3D projection view, both the soybean vegetative meristem and the tomato vegetative meristem form the dome like structure, however, the shape of the soybean vegetative meristem is different from that of the tomato meristem, and the organization and patterns of the leaf primordia surrounding these two SAMs are distinct (Figure 3B-C).

Figure 1: Live imaging and analyzing the inflorescence SAM of Arabidopsis. A and B. optical orthogonal (Orth) and transverse (Trans) section views of middle plane of the same SAM, PI (propidium iodide) stain (purple). C. a 3D projection of the same SAM, PI stain (gray). D. depth color coding of the 3D projection, with blue indicating the top surface layer and red representing the deepest layer. Cell walls were stained with PI. Scale bars: 20 µm (A, B); 50 µm (C, D). Please click here to view a larger version of this figure.
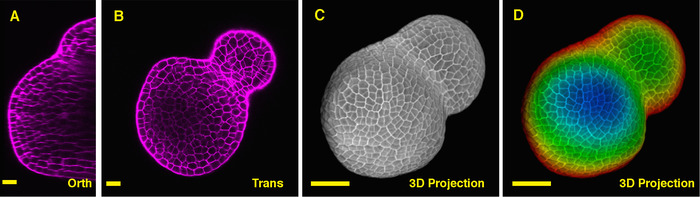
Figure 2: Live imaging and analyzing the vegetative SAM of tomato. A and B. optical orthogonal (Orth) and transverse (Trans) section views of middle plane of the same SAM. C. a 3D projection of the same SAM. D. depth color coding of the 3D projection, with blue indicating the top surface layer and red representing the deepest layer. Cell walls were stained with PI. Scale bars: 20 µm (A, B); 50 µm (C, D). Please click here to view a larger version of this figure.
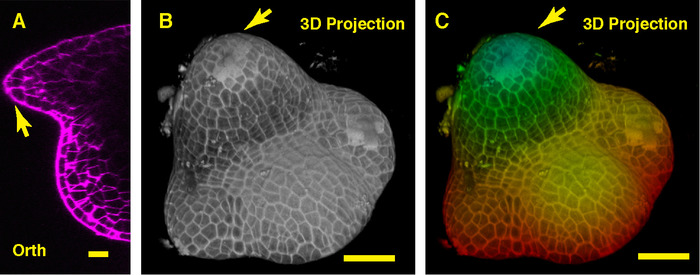
Figure 3: Live imaging and analyzing the vegetative SAM of soybean. A. an optical orthogonal (Orth) section view of the middle plane of the vegetative SAM. B. a 3D projection of the same SAM. C. depth color coding of the 3D projection, with blue indicating the top surface layer and red representing the deepest layer. Cell walls were stained with PI. Scale bars: 20 µm (A); 50 µm (B, C). Arrow: leaf primordium. Please click here to view a larger version of this figure.
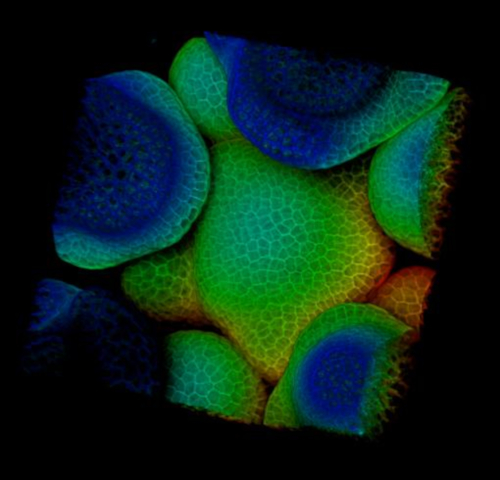
Video 1: 3D rotation of the Arabidopsis inflorescence meristem in Figure 1. Please click here to view this video. (Right-click to download.)
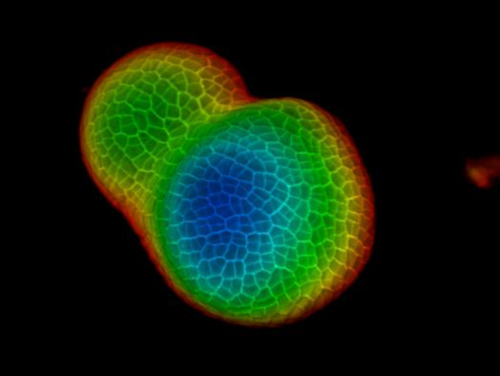
Video 2: 3D rotation of the tomato vegetative meristem in Figure 2. Please click here to view this video. (Right-click to download.)
