This protocol describes in detail a method for protein extraction and digestion followed by phosphopeptide enrichment and subsequent MS analysis (Figure 1). The compositions of all the buffers and solutions that are used in this protocol are listed in Table 1. The sequential use of Lys-C and trypsin provides an efficient digestion. A Coomassie-stained gel of pre-digested lysate confirms the presence of proteins, while staining of post-digested lysate confirms the complete digestion (Figure 2A). For a complete digestion, no bands should appear above 15 kDa, except the 30 kDa and 23.3 kDa bands for Lys-C and trypsin, respectively. The addition of Lys-C also reduces the number of missed cleavages (Figure 2B). Because pY peptides represent only 2% of the phosphoproteome28, immunoprecipitation of the pY peptides using a pY-specific antibody is the first step of pY peptide enrichment. The resulting supernatant becomes the input for pST peptide enrichment. The pY immunoprecipitation effectively separates pY peptides from pST peptides where on average 85% of the phosphopeptides identified from the pY preparation are pY (Figure 3A) and over 99% of the phosphopeptides identified from the pST preparation are pST (Figure 3B). Titanium dioxide is used to enrich for phosphopeptides in both preparations. The expected percentage of peptides in the MS-ready preparation that are phosphorylated is between 30 – 50% (Figure 4A). The variability in the phosphopeptide enrichment percentage may be greater in the pY preparation as a result of there being many fewer pY peptides than pST peptides. In terms of phosphopeptide species, the majority of the phosphopeptides detected have a single or double phosphoryl group (Figure 4B).
After performing mass spectrometry, the MS raw files are loaded into an MS analysis software. The parameter settings used in the experiment are listed in Table 3 but will vary from software to software and may vary from version to version. The parameters that are not listed were left as default, including an FDR cutoff of 1% for peptide-spectrum matching (PSM) with a minimum Andromeda score of 40 for the identification of modified peptides27. Setting a localization probability cutoff of greater than 0.75 filters out approximately 5% of the pY peptides and 15% and 34% of the pS and pT peptides, respectively (Figure 5A). After applying these filters, the expected number of phosphopeptide identifications at the end of the MS analysis is approximately 300 pY peptides (for 5 mg of the starting protein) and about 7,500 pS peptides and 640 pT peptides (for 2.5 mg of the starting peptide amount) from the respective enrichment preparations (Figure 5B). The number of replicates and the variability of the phosphopeptide signal intensity determines adequate powering for statistical comparisons. In four separate experiments with groups containing either biological duplicates or triplicates, the percent coefficients of variation (%CV) for detected phosphopeptides were calculated. Distributions of lower variability (e.g., pST groups 1 – 5 in Figure 5C) indicate that the sample collection, preparation, and mass spectrometry runs were consistent. On the other hand, distributions of higher variability (e.g., pST group 6 in Figure 5C) indicates noisier data that would require larger fold-changes to detect significant differences in downstream differential analyses.
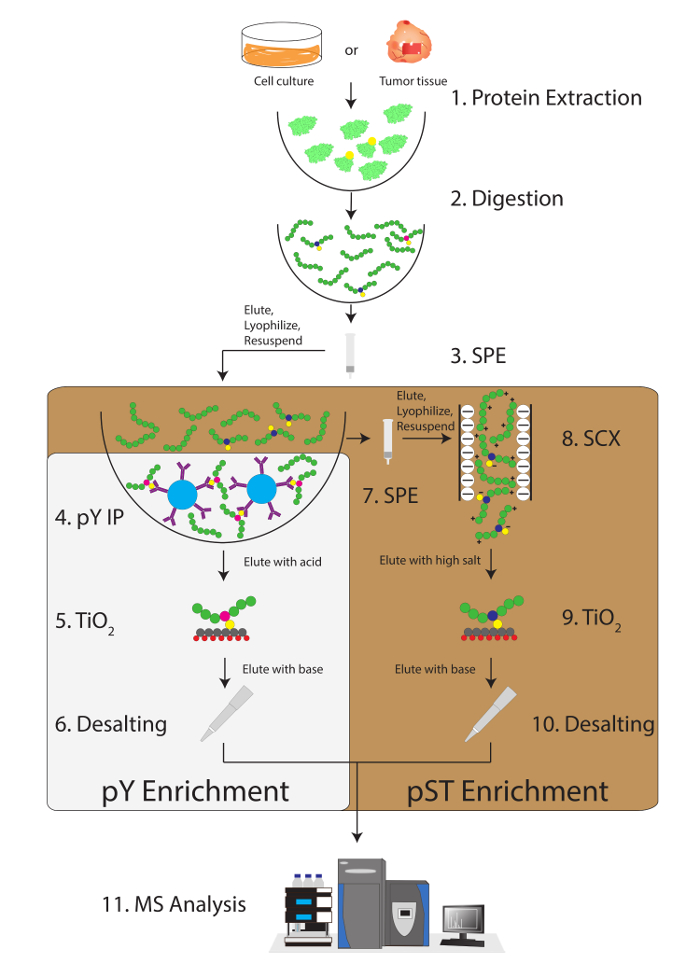
Figure 1: Workflow diagram. Proteins from samples are extracted and digested. Peptides are extracted by solid phase extraction (SPE), and phosphotyrosine (pY) peptides are immunoprecipitated. In parallel, the phosphoserine/threonine (pST) peptides are enriched from the supernatant in the pY immunoprecipitation step. Strong cation exchange (SCX) is performed on the supernatant to remove highly charged peptides to reduce the ion suppression12. Both preparations undergo phosphopeptide enrichment via titanium dioxide (TiO2). After sample cleanup, liquid chromatography-tandem mass spectrometry (LC-MS/MS) is performed to measure the phosphopeptide abundance. The raw data is then loaded into an MS analysis software to identify phosphopeptides. Please click here to view a larger version of this figure.
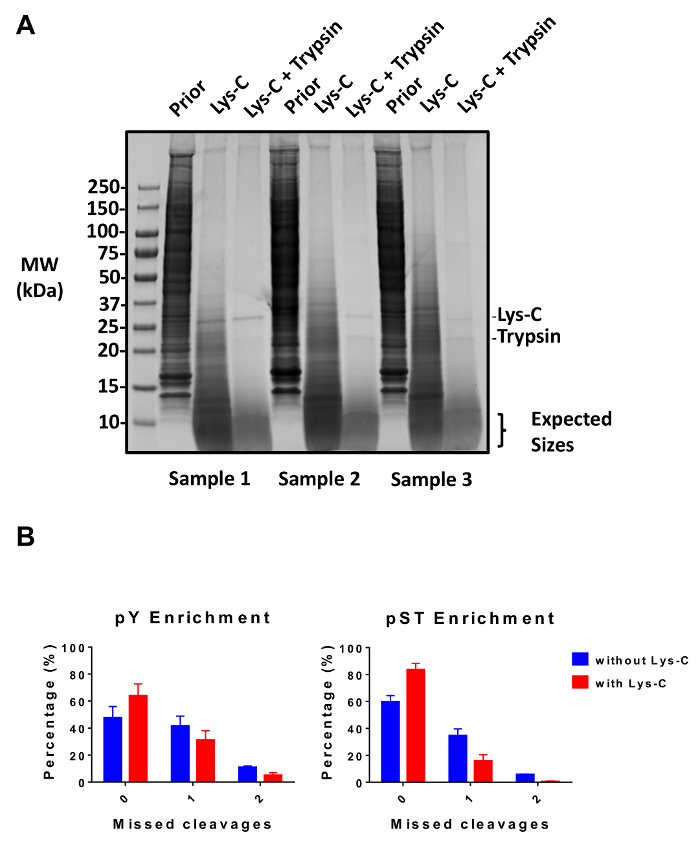
Figure 2: Evaluation of digestion. (A) Three samples with 12.5 µg of lysate pre-digestion, post-Lys-C digestion, and post-trypsin digestion are shown. A Coomassie gel-stain test shows a clean digestion after sequential use of Lys-C and trypsin. The molecular weight (MW) size markers are in kilodaltons (kDa). (B) A reduction in missed cleavages is observed after Lys-C was added to the protocol. The percentage of phosphopeptides without missed cleavages increased from 48% to 64% and from 60% to 84% on average for pY and pST enrichment preparations, respectively. The graphs summarize the data obtained from two experiments performed without Lys-C and five experiments performed with Lys-C. The error bars are standard deviations representing 38 pY and 38 pST samples from 2 separate experiments (without Lys-C) and 62 pY and 60 pST samples from 5 separate experiments (with Lys-C). Please click here to view a larger version of this figure.
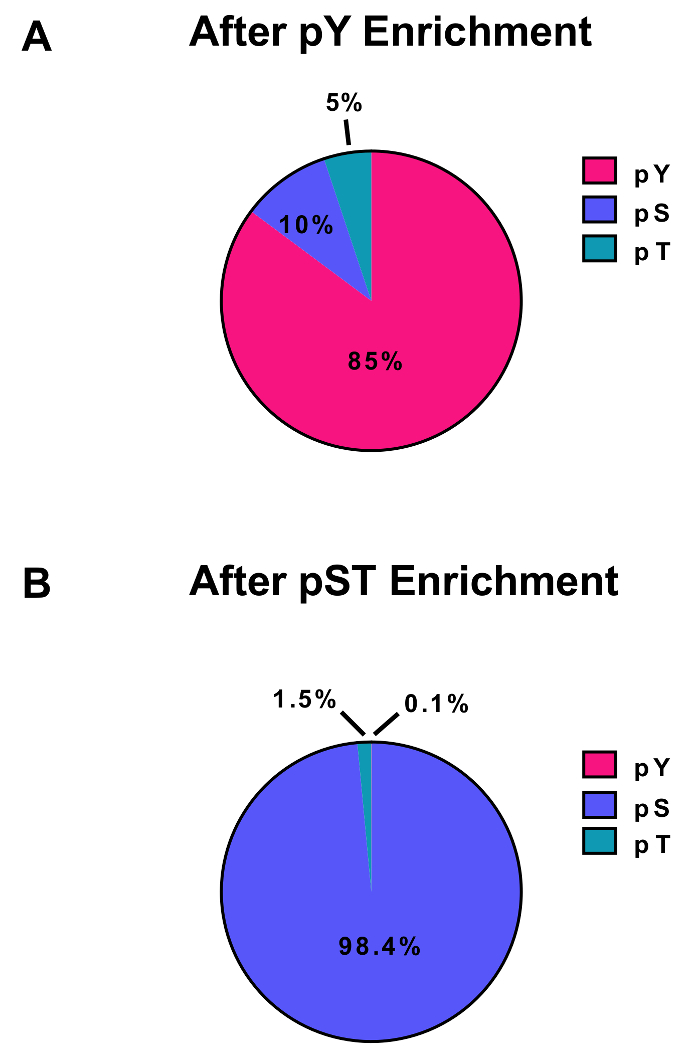
Figure 3: Enrichment of pY and pST phosphopeptides. These panels show the percentages of pSTY phosphopeptides from either (A) the pY or (B) the pST enrichment preparations. The pY enrichment by pY immunoprecipitation and titanium dioxide resulted in 85% phosphopeptides being for pY peptides, while only 0.1% of the phosphopeptides in the pST enrichment are pY. These values were drawn from examining the Phospho (STY)Sites.txt file of one representative experiment after filtering out contaminants, reverse sequences, and phosphopeptides with localization probabilities less than 0.75. Please click here to view a larger version of this figure.

Figure 4: Phosphopeptide enrichment with titanium dioxide. (A) The percentage of detected phosphopeptides (relative to total peptides) from samples in four separate experiments is shown. (B) This panel shows the average composition of mono-, double-, and multi-phosphorylated peptides in four separate experiments. The error bars in panel A are standard deviations. Please click here to view a larger version of this figure.
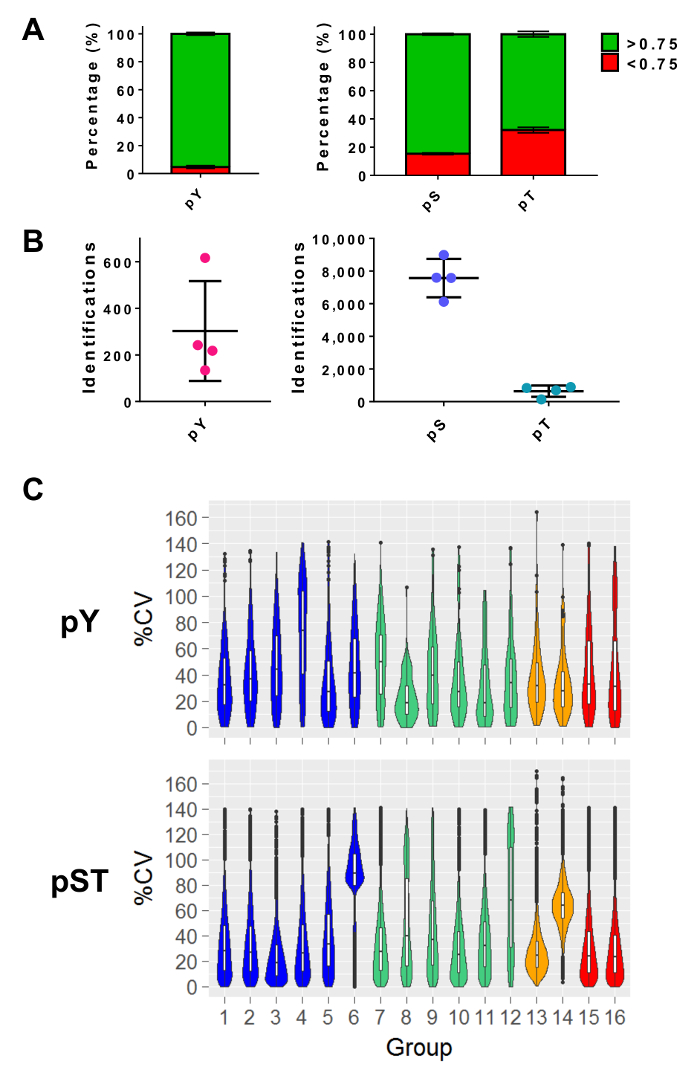
Figure 5: Expected phosphoresidue identifications. (A) This panel shows the phosphorylation localization probabilities of IDs from pY enrichment (left) and pST enrichment (right). The mean percentage of IDs that meet the > 0.75 probability cutoff is 93%, 75%, and 52% for pY, pS, and pT, respectively. (B) The mean number of IDs with a >0.75 localization probability is 300 for pY, 7,500 for pS, and 640 for pT. (C) This panel shows violin plots of the percent coefficient of variation (%CV) of the phosphopeptides. An evaluation of %CV was only performed if a signal intensity value was detected in each biological replicate or triplicate group. Data was taken from four separate experiments. The error bars in panels A and B are standard deviations from 34 pY and 34 pST samples from 4 separate experiments. Please click here to view a larger version of this figure.
| Buffer | Volume | Composition | |
| 6 M guanidinium chloride lysis buffer | 50 mL | 6 M guanidinium chloride, 100 mM tris pH 8.5, 10 mM tris (2-carboxyethyl) phosphine, 40 mM chloroacetamide, 2 mM sodium orthovanadate, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 500 mg n-octyl-glycoside, ultra-pure water to volume | |
| 100 mM sodium pyrophosphate | 50 mL | 2.23 g sodium pyrophosphate decahydrate, ultra-pure water to volume | |
| 1M β-glycerophosphate | 50 mL | 15.31 g β-glycerophosphate, ultra-pure water to volume | |
| 5% trifluoroacetic acid | 20 mL | Add 1 mL of 100% trifluoroacetic acid into 19 mL ultra-pure water | |
| 0.1% trifluoroacetic acid | 250 mL | Add 5 mL 5% trifluoroacetic acid to 245 mL ultra-pure water | |
| pY elution buffer | 250 mL | 0.1% trifluoroacetic acid, 40% acetonitrile, ultra-pure water to volume | |
| pST elution buffer | 250 mL | 0.1% trifluoroacetic acid, 50% acetonitrile, ultra-pure water to volume | |
| IP binding buffer | 200 mL | 50 mM tris pH 7.4, 50 mM sodium chloride, ultra-pure water to volume | |
| 25 mM ammonium bicarbonate, pH 7.5 | 10 mL | Dissolve 19.7 mg into 10 mL sterile ultra-pure water, pH to 7.5 with 1 N hydrochloric acid (~10-15 µL/10 ml solution), make fresh | |
| 1M phosphate buffer, pH 7 | 1,000 mL | 423 mL 1 M sodium dihydrogen phosphate, 577 mL 1 M sodium hydrogen phosphate | |
| Equilibration buffer | 14 mL | 6.3 mL acetonitrile, 280 µL 5% trifluoroacetic acid, 1740 µL lactic acid, 5.68 mL ultra-pure water | |
| Rinsing buffer | 20 mL | 9 mL acetonitrile, 400 µL 5% trifluoroacetic acid, 10.6 mL ultra-pure water | |
| Mass spectrometry solution | 10 mL | 500 µL acetonitrile, 200 µL 5% trifluoroacetic acid, 9.3 mL ultra-pure water | |
| Buffer A | 250 mL | 5 mM monopotassium phosphate (pH 2.65), 30% acetonitrile, 5 mM potassium chloride,ultra-pure water to volume | |
| Buffer B | 250 mL | 5 mM monopotassium phosphate (pH 2.65), 30% acetonitrile, 350 mM potassium chloride, ultra-pure water to volume | |
| 0.9% ammonium hydroxide | 10 mL | 300 μL 29.42% ammonium hydroxide, 9.7 mL ultra-pure water | |
Table 1: Buffers and solutions. This table shows the compositions of the buffers and solutions used in this protocol.
| LC-MS/MS Settings | ||
| Parameter | pY Setting | pST Setting |
| Sample loading (µL) | 5 | |
| Loading flow rate (µL/min) | 5 | |
| Gradient flow rate (nL/min) | 300 | |
| Linear gradient (percentage 0.16% formic acid, 80% ACN in 0.2% formic acid) | 4 – 15% for 5 min | 4 – 15% for 30 min |
| 15 – 50% for 40 min | 15 – 25% for 40 min | |
| 50 – 90% for 5 min | 25 – 50% for 44 min | |
| 50 – 90% for 11 min | ||
| Full scan resolution | 120,000 | |
| Number of most intense ions selected | 20 | |
| Relative collision energy (%) (HCD) | 27 | |
| Dynamic exclusion (s) | 20 | |
Table 2: LC-MS settings. This is an example of LC-MS settings in a typical shotgun phosphoproteomic experiment. The samples were loaded on to a trap column. The trap was brought in-line with an analytical column. These settings were optimized for using the LC-MS system listed in the Table of Materials and Reagents. These settings would need to be adjusted for other LC-MS systems.
| MaxQuant Parameter Settings | ||
| Setting | Action | |
| Group-Specific Parameters | ||
| Tipo | Tipo | Select Standard |
| Multiplicity | Set to 1 | |
| Digestion Mode | Enzyme | Select Trypsin/P |
| Max. missed cleavages | Set to 2 | |
| Modifications | Variable modifications | Add Phospho (STY) |
| Label-free quantification | Label-free quantification | Select LFQ |
| LFQ min. ratio count | Set to 1 | |
| Fast LFQ | Check off | |
| Miscellaneous | Re-quantify | Check off |
| Global Parameters | ||
| Sequences | FASTA files | Select fasta file downloaded from UniProt |
| Fixed modifications | Add Carbamidomethyl (C) | |
| Adv. Identification | Match between runs | Check off |
| Match time window | Set to 5 min | |
| Alignment time window | Set to 20 min | |
| Match unidentified features | Check off | |
| Protein quantification | Min. ratio count | Set to 1 |
| Folder locations | Modify accordingly | |
Table 3: MS analysis software settings. In MaxQuant, the group-specific and global parameters in this table were selected or adjusted. All other parameters remained at default. These experiments were conducted using version 1.5.3.30. The parameters may vary from version to version and from software to software.
