Improved Polymerase Chain Reaction-restriction Fragment Length Polymorphism Genotyping of Toxic Pufferfish by Liquid Chromatography/Mass Spectrometry
Summary
An improved polymerase chain reaction-restriction fragment length polymorphism method for genotyping pufferfish species by liquid chromatography/mass spectrometry is described. A reverse-phase silica monolith column is employed for separating digested amplicons. This method can elucidate the monoisotopic masses of oligonucleotides, which is useful for identifying base composition.
Abstract
An improved version of a polymerase chain reaction (PCR)-restriction fragment length polymorphism (RFLP) method for genotyping toxic pufferfish species by liquid chromatography/electrospray ionization mass spectrometry (LC/ESI-MS) is described. DNA extraction is carried out using a silica membrane-based DNA extraction kit. After the PCR amplification using a detergent-free PCR buffer, restriction enzymes are added to the solution without purifying the reaction solution. A reverse-phase silica monolith column and a Fourier transform high resolution mass spectrometer having a modified Kingdon trap analyzer are employed for separation and detection, respectively. The mobile phase, consisting of 400 mM 1,1,1,3,3,3-hexafluoro-2-propanol, 15 mM triethylamine (pH 7.9) and methanol, is delivered at a flow rate of 0.4 ml/min. The cycle time for LC/ESI-MS analysis is 8 min including equilibration of the column. Deconvolution software having an isotope distribution model of the oligonucleotide is used to calculate the corresponding monoisotopic mass from the mass spectrum. For analysis of oligonucleotides (range 26-79 nucleotides), mass accuracy was 0.62 ± 0.74 ppm (n = 280) and excellent accuracy and precision were sustained for 180 hr without use of a lock mass standard.
Introduction
Mass spectrometry (MS) is an accepted technology for rapid and reliable identification of nucleic acids, matrix-assisted laser desorption/ionization (MALDI) and electrospray ionization (ESI) being used for ionization1,2. The MALDI technique is typically combined with a time-of-flight (TOF) analyzer; however, the application of MALDI-TOF MS is limited to short oligonucleotides (~25 nucleotides (nt)) owing to the subsequent fragmentation, adduct formation and low ionization efficiency1,2. In contrast, ESI is applicable to longer oligonucleotides (>100 nt), but many charge states of multiple charged ions ([M−nH]n−) are produced simultaneously from biomacromolecules and, therefore, the mass of the analyte can exceed the upper limit of the m/z range of the spectrometer. This requires the interpretation of the convoluted spectra, i.e., transformation of a charge state series into the corresponding mass via deconvolution.
In the case of mass measurement of a small molecule, the peak of the monoisotopic mass, i.e., the mass of the molecule containing only the most common isotope of each element is the most abundant and observed naturally3. As the molecular weight increases, the isotopic distribution shifts to higher m/z and the monoisotopic peak becomes obscured by baseline noise3-5. Once the monoisotopic peak is no longer detectable for masses greater than 10 kDa3, the average molecular mass is used for measurement rather than the monoisotopic mass5. In such cases, the isotopic distribution in which each of the individual peaks is separated by 1 Da can only be observed when a high resolution mass analyzer such as a TOF, a Fourier transform modified Kingdon trap analyzer6, or a Fourier transform ion cyclotron resonance analyzer is used for analysis. However, the most abundant peak is sometimes not consistent with the average molecular mass5. These problems can lead to difficulty for accurately determining analytes.
Given the variation in the natural abundance of stable isotopes and the difficulties in the determining the average molecular mass, measurement of the monoisotopic mass is ideal for mass characterization of biomolecules3,4. In practice, whether a monoisotopic peak can be observed or not, the monoisotopic mass can be estimated by comparing the pattern of the observed isotopic distribution to the theoretical one calculated from a model analyte4,7-10. The fitting algorithm8 is now incorporated into proprietary software.
In the context of ESI-MS, dissociation of a DNA duplex, purification and desalting are required for direct measurement to avoid the failure of ionization due to ion suppression and adduct formation2,10-14. These procedures are troublesome, however, fully automated analytical systems have been developed commercially involving polymerase chain reaction (PCR), sample preparation and ESI-MS for the detection of pathogens15-20. Another approach is introducing liquid chromatography (LC) for separation. LC provides an on-line separation of the analytes from coexisting substances and does not require a laborious sample preparation prior to ionization21,22. However, separation of nucleic acids using a MS-compatible mobile phase is more difficult than for most other compounds such as drugs, peptides and proteins owing to the polyanionic nature of the nucleotides. The most successful examples of LC/ESI-MS involve the use of ion-pair reverse-phase LC. A mobile phase of 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP)-triethylamine (TEA)-methanol was initially proposed by Apffel et al. for separating and detecting short oligonucleotides23. Application of LC/ESI-MS genotyping for differentiation of pathogen species, single-nucleotide polymorphisms and short tandem repeats has been reported using a capillary polymer-monolith column that can separate longer oligonucleotides1,21,24-33.
In Japan and the United States, serious food poisoning has occurred due to misidentification and inappropriate preparation of pufferfish, and this is despite the fact that the distribution and preparation of pufferfish is strictly controlled by food safety legislation34,35. Moreover, an intentional homicide using pufferfish extract did occur in Japan36. Therefore, differentiation of pufferfish species is required from both public health and forensic investigative standpoints. Additionally, because Takifugu rubripes is far more expensive than other kinds of pufferfish, a differentiation capability is also needed in the context of food fraud investigations.
Here, a detailed method for determining the monoisotopic mass of a PCR product by LC/ESI-MS using a reverse-phase silica monolith column and a high resolution mass spectrometer is described. Specifically, the approach has been developed to permit differentiation of toxic pufferfish species based on the use of the PCR restriction fragment length polymorphism (RFLP) method37, which is the first example for species differentiation of animals using MS.
Protocol
1. DNA Extraction
Note: Use a separate room for DNA extraction and post-PCR examinations such as gel electrophoresis and LC/ESI-MS. Add ethanol to wash buffer 1 (containing guanidine hydrochloride) and wash buffer 2 (not containing guanidine hydrochloride) according to the DNA extraction kit protocol. Fish samples were obtained from fish wholesalers and retail markets in Japan.
- Place 30-50 mg of fish tissue in a 0.5 ml or 1.5 ml microcentrifuge tube. Squash the tissue using a micro-spatula (except for fin and skin).
Note: In the case of fin, use a 0.5 ml tube for soaking. - Add 180 µl of the lysis buffer and 40 µl of proteinase K and vortex briefly. Incubate at 56 °C on a block heater for 2 hr or overnight. Mix by briefly vortexing every 15 min during the incubation.
Note: Complete lysis is not necessary. - Centrifuge for 5 min at 13,000 x g. After centrifugation, if a small amount of oil exists on the surface in the case of a fatty sample such as liver, discard it. Transfer supernatant to a new 1.5 ml microcentrifuge tube.
- Add 200 µl of the chaotropic buffer containing guanidine hydrochloride and mix by vortexing. Add 200 µl of ethanol (99.5%) and mix by vortexing again.
- Transfer the mixture into the spin column placed in a 2 ml collection tube. Centrifuge for 1 min at 13,000 x g. Discard the flow-through.
Caution: Do not add bleach to the waste because guanidine can form highly reactive compounds with bleach. - Add 500 µl of the wash buffer 1 (containing guanidine hydrochloride) and centrifuge for 1 min at 13,000 x g. Discard the flow through and the collection tube.
- Place the spin column in a new 2 ml collection tube provided with the kit. Add 500 µl of the wash buffer 2 and centrifuge for 3 min at 20,000 x g. Discard the flow-through and the collection tube. Transfer the spin column to a new 1.5 ml microcentrifuge tube.
- Pipet 200 µl of the elution buffer to the center of the spin column membrane. After 1 min, centrifuge for 1 min at 6,000 x g.
- Pipet 50 µl of the eluate to a disposable UV cuvette and measure the UV spectrum of the eluate at 220-300 nm and the absorbance at 260 nm using a spectrophotometer.
- Verify the UV spectrum of DNA with the peak at 260 nm. Calculate the approximate DNA concentration using the simple equation "DNA concentration (ng/µl) = absorbance at 260 nm x 50".
Note: Typical DNA concentration extracted from the fins of pufferfish is 63 ± 30 ng/µl (n = 20). - If the DNA concentration is higher than 10 ng/µl, dilute the DNA samples with Tris-EDTA (TE) buffer (pH 8) to 5 ng/µl.
- Store the sample at −20 °C or proceed to PCR amplification (section 2).
2. PCR
- Set up 25 µl PCR for each extracted DNA sample, positive control (0.2 µM synthesized oligonucleotide having the target sequence in TE buffer pH 8.0) and negative control (ultrapurified water instead of DNA sample) on ice as per Tables 1 and 2 on a clean bench to avoid contamination.
Note: For easy operation, make sufficient amount of cocktail containing all reagents without template DNA and dispense it. Use DNA polymerase having proofreading activity to avoid the addition of 3' A overhangs to the PCR product. - Carry out PCR amplification on a thermal cycler using the cycle program shown in Table 3.
3. Enzymatic Digestion
- Add 2 µl of the solution containing 100 mM Tris-HCl (pH 7.5), 100 mM MgCl2, 10 mM dithiothreitol and 500 mM NaCl to the PCR solution.
- Add 1 µl of each restriction enzyme (Dra I and Msp I) and mix gently. Incubate for 30 min at 37 °C on the thermal cycler.
- Incubate for 5 min at 72 °C to activate the remaining DNA polymerase, which fills the sticky end generated by Msp I.
- Optional: Analyze each 2.5 µl of the reaction solution by polyacrylamide gel electrophoresis using a cross link ratio (%C, percentage of bisacrylamide) of 3.33 (wt %) and a polyacrylamide gel concentration (%T) of 15 (% w/v)38. Stain the gel using the fluorescent DNA stain and observe double-strand DNA on a blue light transilluminator using an amber screen.
- Filter the reaction solution with a 0.2-0.5 µm centrifugal filter device to prevent clogging, which would damage the analytical column.
- Transfer the filtrate to a tapered polypropylene vial and store at -20 °C until analysis.
4. LC/ESI-MS Analysis
- Weigh 67.2 g (ca. 42 ml) of HFIP and 1.52 g (ca. 2.0 ml) of TEA in a 1 L bottle. Add 955 ml of ultrapurified water (LC/MS grade) and stir using a magnetic stirrer until the reagents are completely dissolved. Take an aliquot of the solution and check the pH (between 7.85 and 7.95; ideally pH = 7.9) using a pH meter.
Note: Do not return the aliquot to the bottle to avoid contamination by metal ions. If the pH is not 7.9, add a small amount of either HFIP or TEA to adjust the pH and measure the pH again. - Connect the bottle to line A of the liquid chromatograph with cooling of the bottle using a direct cooling portable refrigerator (for household use) to prevent vaporization. Connect another bottle filled with methanol to line B.
- Connect the guard column and the analytical column (2.0 x 50 mm, mesopore size = 30 nm) to the liquid chromatograph. Start flowing the initial mobile phase (95% A and 5% B) to equilibrate the columns.
- Prepare 0.5 mg/ml sodium trifluoroacetate (pH 3.5) as a calibrant39.
- Weigh 50 mg (ca. 34 µl) of trifluoroacetic acid in a beaker.
- Add 30 ml of ultrapurified water and titrate to pH 3.5 with 10 mM sodium hydroxide with the aid of a pH meter.
- Fill up to 50 ml with ultrapurified water. Add 50 ml of acetonitrile (LC/MS grade) and mix.
- Take a portion of the working solution and store it at room temperature. Store the rest of the solution at 4 °C.
- Reduce the nitrogen pressure of the C-trap as follows.
Note: Recent Fourier transform mass spectrometers, which are suitable for intact protein analysis, have control software to control pressure.- Remove the left top cover of the mass spectrometer. The C-trap valve is at the corner of the left foreground.
- Check high vacuum pressure value in the instrument status window of the control software.
Note: The value is typically, 3 x 10−8 bar. - Pull the knob to unlock and turn the knob anticlockwise very slowly to reduce the high vacuum pressure to 1/3 (typically, 1 x 10−8 bar). The indicated pressure should change gradually in a delayed manner. Ignore the warning message about low pressure. Push the knob to lock.
- Restore the top cover for safety.
- Calibrate the mass spectrometer using sodium trifluoroacetate39.
Note: Daily mass calibration can be replaced by this protocol, however, the recommended calibration suggested by the manufacturer should have been completed prior to this calibration.- Set scan parameters and heated ESI source parameters in the instrument control box of the tuning software as follows: scan type full MS; scan range m/z 500-4,000; fragmentation (in-source collision induced dissociation (CID) voltage 60 eV; polarity negative; auto gain control (AGC) target 1 x 106, maximum inject time 50 msec; sheath gas flow rate 5; aux gas flow rate 0; sweep gas flow rate 0; spray voltage 3 kV; capillary temperature 320 °C; S-lens radiofrequency (RF) level 100; heater temperature 30 °C.
- Input the list of negative ions to be monitored as m/z 792.85908, 1064.80870, 1336.75832, 1608.70794, 1880.65755, 2152.60717, 2424.55679, 2696.50640, 2968.45602, 3376.38045.
- Move the heated ESI probe closer to the interface of the mass spectrometer (position B). Connect the probe and a syringe with a tube.
- Infuse the calibrant constantly at a flow rate of 10 µl/min using the embedded syringe pump. Check only the 6 lower mass ions (m/z 792.85908-2152.60717) in the customized calibration table.
- Proceed with calibration after the intensity of the signals are stabilized. After the calibration, check only the six higher mass ions (m/z 1880.65755-3376.38045) and proceed with calibration. Avoid calibrating using all ions at once to avoid failure.
- Set scan parameters and heated ESI source parameters in the instrument control box of the tuning software as follows: scan type full MS; scan range m/z 500-4,000; fragmentation (in-source collision induced dissociation (CID) voltage 60 eV; polarity negative; auto gain control (AGC) target 1 x 106, maximum inject time 50 msec; sheath gas flow rate 5; aux gas flow rate 0; sweep gas flow rate 0; spray voltage 3 kV; capillary temperature 320 °C; S-lens radiofrequency (RF) level 100; heater temperature 30 °C.
- Move the probe to the appropriate position for a flow rate of 0.4 ml/min (position D) and connect to the liquid chromatograph with an inert metal tube.
- Set the heated ESI source parameters in the instrument control box of the tuning software as follows: sheath gas flow rate 50; aux gas flow rate 15; sweep gas flow rate 1; spray voltage 2.5 kV; capillary temperature 350 °C; S-lens RF level 100; heater temperature 350 °C.
- Set acquisition parameters of the mass spectrometer in the instrumental setup window as follows: method duration 8 min; divert valve, 3.5-6 min to mass spectrometer, 0-3.5 and 6-8 min to waste; runtime 3.5 to 6 min; polarity negative; in source CID voltage 15.0 eV; microscans 3; nominal resolution power (at m/z 200) 140,000; AGC target 1 x 106; maximum ion time 50 msec; number of scans 1; scan range m/z 700-3,500; spectrum data type profile.
- Set parameters of liquid chromatograph as follows: column oven temperature 20 °C; sample tray temperature 10 °C; gradient program 0-0.5 min 5% B, 0.5-1 min 5-30% B, 1-3.5 min 30-40% B, 3.5-5 min 40-98% B, 5-6 min 98% B, 6-6.05 min 98-5% B, 6.05-8 min 5% B; flow rate 0.4 ml/min.
- Purge bubbles by tapping the bottoms of the vials. Set sample vials on the sample rack and inject 1.0 µl of a sample using the autosampler to start analysis.
- Open raw data files with the browser software and confirm that all the acquisitions are successfully finished, including the positive and negative controls.
Note: Typically, two or three peaks would be observed in the total ion current chromatogram at a retention time of 4-5.5 min when pufferfish DNA is successfully amplified and digested.
5. Deconvolution and Interpretation
- Start the deconvolution software. Select experiment type "auto extract (isotopically resolved)".
- Edit a method using the following parameters: output mass M; S/N threshold 3, relative abundance threshold 0; negative charge yes; calculate extracted ion current chromatogram (XIC) yes; m/z range 700-3,500; charge carrier H+; minimum number of detected charge 3; isotope ratio nucleotide; fit factor 80%; remainder threshold 0%; consider overlaps yes; charge range 3-50; minimum intensity 1; expected intensity error 3.
- Save as a new method and select it.
- Select the directory where the raw data files exist. Select the raw data files to be analyzed and click 'Add to Queue' button.
- Click 'Run' button at 'Run Queue' tab to start deconvolution.
- After the queue is completed, select a row and click 'Open Result' in the upper caption to obtain the results of deconvolution.
- Interpret the results from the monoisotopic masses derived from the peaks at a retention time of 4-5.5 min using Table 4.
Representative Results
Three commercially-available columns were evaluated for the separation of long oligonucleotides at flow rates of 100-400 µl/min. A wide-pore octadecyl carbon chain (C18)-bonded particulate silica column (a), a commercially-available poly(styrene-divinylbenzene) (PS-DVB) monolith column (b) and a C18-bonded silica monolith column (c) were compared (Figure 1). Three pairs of the DNA duplexes (26, 37 and 53 bp) were separated using all the columns, and the C18-bonded silica monolith column was used in subsequent studies.
Representative results for the analysis of a DNA sample extracted from the muscle of T. rubripes is shown in Figure 2. Isotopically resolved peaks, as shown in Figure 2a, are required for successfully calculating the monoisotopic mass. The theoretical and measured monoisotopic mass of T. rubripes are shown in Figure 2b. Because the digestion with the endonucleases is performed just after PCR amplification without purification, the 3ʹ-sticky end generated by the Msp I endonuclease is filled up with the remaining DNA polymerase. According to the analysis of amplicons derived from the synthetic DNA templates, mass accuracy ranged from -2.48 to 2.40 ppm (0.62 ± 0.74 ppm on average, n = 280). Thus, a mass tolerance of 3 ppm was used for differentiating species (Table 4). Using the table, all of the pufferfish samples obtained from markets and stores were properly differentiated as specific species37.
According to the periodic analysis of the sample obtained from the synthetic DNA template of T. chrysops, all the monoisotopic masses of the analytes were successfully determined within ± 3 ppm for at least 180 hr without any mass calibration (Figure 3). This means that mass calibration would be required on a weekly basis.
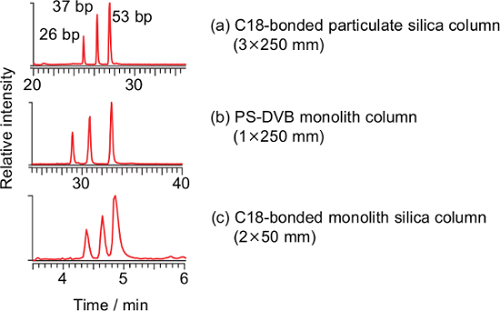
Figure 1: Total ion current chromatograms of the PCR-RFLP product derived from the synthesized DNA template of T. pardalis using a C18-bonded particulate silica column (a, 3×250 mm, particle size 3 µm), a poly(styrene-divinylbenzene) (PS-DVB) monolith column (b, 1.0×250 mm) and a C18-bonded monolith silica column (c, present method). Conditions for (a): flow rate 0.2 ml/min; ratio of methanol, 0-2 min 5%, 2-5 min 5%-25%, 5-20 min 25%-40%, 20-35 min 40%-98%, 35-50 min 98%, 50-51 min 98%-5%, 51-65 min 5%. Conditions for (b): flow rate 0.1 ml/min; ratio of methanol, 0-2 min 5%, 2-5 min 5%-25%, 5-35 min 25%-40%, 35-40 min 40%-98%, 40-55 min 98%, 55-56 min 98%-5%, 56-70 min 5%. The temperature for both columns was 20 °C. Please click here to view a larger version of this figure.
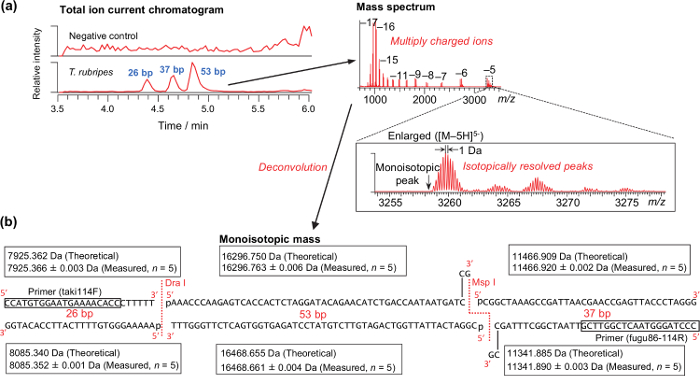
Figure 2: Representative results for analyzing raw muscle of T. rubripes. (a) Typical total ion current chromatograms and mass spectrum. (b) Theoretical and measured monoisotopic masses of the PCR-RFLP products derived from the synthesized DNA template of T. rubripes. Please click here to view a larger version of this figure.
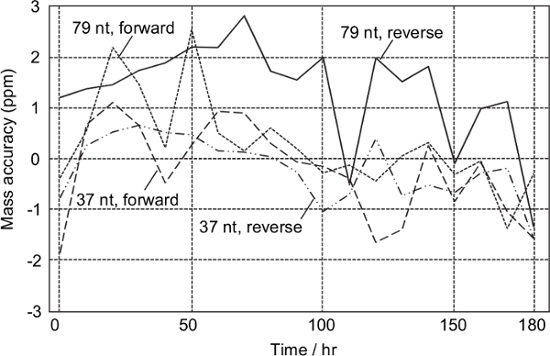
Figure 3: Stability test. Digested amplicons derived from the synthetic DNA of T. chrysops were analyzed every 10 hr. Mass calibration was not performed during the test. Please click here to view a larger version of this figure.
| Name | Sequence |
| lago86F | 5ʹ-CCATGTGGAATGAAAACACC-3ʹ |
| taki114F | 5ʹ-AAAAACAAGAGCCACAGCTCTAA-3ʹ |
| fugu86-114R | 5ʹ-CCCTAGGGTAACTCGGTTCG-3ʹ |
Table 1: PCR primers.
| PCR reagent | Volume used | Final concentration |
| Ultrapurified water | 15.75 μl | |
| Detergent-free 5x Buffer | 5.0 μl | 1x |
| dNTP mix (10 mM each) | 0.5 μl | 0.2 mM |
| Primer mix (10 μM each of lago86F, taki114F and fugu86-114R in TE buffer pH 8.0) | 1.0 μl | 0.4 μM each |
| DNA polymerase (2.5 unit/μl) | 0.25 μl | 0.1 unit/μl |
| Template DNA in TE buffer pH 8.0 (5.0–10 ng/μl for extracted sample, 0.2 μM for synthetic DNA as positive control) | 2.5 μl | 0.5-1.0 ng/μl for extracted DNA and 20 nM for synthetic DNA |
| Total: 25 μl |
Table 2: Components of a 25 µl scale PCR.
| Cycle | Condition | Function |
| 1 | 2 min at 95 °C | Initial denaturation |
| 2 | 30 s at 95 °C | Denaturation |
| 3 | 30 s at 56 °C | Annealing |
| 4 | 30 s at 72 °C | Elongation |
| 5 | Repeat 2-4 (totally 30 cycles) | |
| 6 | 7 min at 72 °C | Final Elongation |
| 7 | Hold at 12 °C until removal of the sample |
Table 3: PCR program.
| Peak number at retention time of 4.0-5.5 min | Monoisotopic mass of the third peak in the range (Da) | Monoisotopic mass of the second peak in the range (Da) | Pufferfish species | ||
| Smaller strand | Larger strand | Smaller strand | Larger strand | ||
| 2 | → | → | 15890.707–15890.803 | 16177.531–16177.629 | L. inermis |
| 15921.702–15921.797 | 16146.537–16146.634 | L. gloveri | |||
| 15930.713–15930.809 | 16137.525–16137.622 | L. lunaris | |||
| 15937.697–15937.792 | 16131.537–16131.634 | L. wheeleri | |||
| 24173.034–24173.179 | 24566.905–24567.053 | T. chrysops | |||
| 3 | 16295.706–16295.804 | 16467.610–16467.709 | 11341.851–11341.919 | 11466.875–11466.944 | T. pardalis T. snyderi T. ocellatus T. xanthopterus T. stictonotus |
| 11654.908–11654.978 | 11770.920–11770.991 | T. niphobles | |||
| 16296.701–16296.799 | 16468.605–16468.704 | → | → | T. rubripes | |
| 16311.701–16311.799 | 16452.610–16452.709 | → | → | T. poecilonotus T. exascurus |
|
| 16320.713–16320.811 | 16443.599–16443.697 | → | → | T. porphyreus T. obscurus |
|
| 16599.751–16599.851 | 16780.667–16780.767 | → | → | T. vermicularis | |
Table 4: Differentiation of pufferfish from monoisotopic masses derived from LC/ESI-MS.

Table 5: Amplified DNA sequence. (a) The sequence is identical to that of the other pufferfish species as described in Table 4. Please click here to view a larger version of this table.
Discussion
To extract DNA, a commercial kit for extracting DNA from blood and tissues is used in accordance with the protocol of the manufacturer with minor modification (amount of proteinase K and centrifugation of the lysis solution). However, any extraction kit can be used as long as cellular DNA can be extracted with appropriate recovery and purity for PCR. This method has been tested using muscle, fin, liver, ovary, and skin37. Fin is especially suitable because of the large surface area, which enables rapid lysis with proteinase K. Fresh, frozen, dried, boiled and baked pufferfish samples were tested successfully37.
The PCR primer set (Table 1) is designed for pufferfish and amplifies the mitochondrial 16S ribosomal RNA gene of pufferfish. The target DNA to amplify is 114-115 bp (genus Takifugu) and 86 bp (genus Lagocephalus) (Table 5). To facilitate method development, synthesized oligonucleotides having the target sequences were used for the reference instead of collecting authentic pufferfish specimens. The primer set can be replaced by another primer set in response to the purpose, however, final DNA length after enzymatic digestion should be less than 100 nt in terms of maintaining the quality of the mass spectrum required for successful deconvolution. Additionally, nitrogen pressure in the C-trap should be reduced when the target oligonucleotide is longer than about 75 nt and the quality of the mass spectrum is insufficient for successful deconvolution. Such limitations can be controlled through recent developments in instrument software, otherwise the valve for the C-trap inside the chassis must be tuned as described in the protocol section. As for sample preparation, use of detergent-free reagents is critical for the subsequent LC in terms of appropriate peak shape, sufficient peak intensity and stable retention time31.
A PS-DVB capillary monolith column21,24-33, a C18 reverse-phase particulate silica column23,40,41 and a hydrophobic interaction chromatography (HILIC) column42 have been used for the LC/ESI-MS analysis of oligonucleotides. Among them, the capillary monolith column is superior to the others in terms of separation capacity, however, the capillary monolith column used in past studies was made in-house and operated at a low flow rate (2 µl/min), which requires instrumentation dedicated to micro LC. To facilitate easy operation, commercially-available columns were evaluated for the separation of long oligonucleotides at higher flow rates (100-400 µl/min). Three pairs of DNA duplexes (26, 37 and 53 bp) were separated using the above-mentioned columns, however, the cycle times of the C18-bonded particulate silica column and the PS-DVB monolith column were 65 and 70 min, respectively, whereas that of the C18-bonded silica monolith column was 8 min (Figure 1). Taking rapid analysis into consideration, the C18-bonded silica monolith column was chosen for our purposes despite the limited separation capacity; however the remaining two columns may be employed when improved separation is required. Theoretically, in the case of a monolith column, there is no interstitial volume and the mobile phase would be forced to flow through the pores of the solid phase while maintaining a consistent path length, thus enabling an efficient separation27. Such processes would be manifested, particularly in the analysis of biomacromolecules such as an oligonucleotide, as the slow diffusion of the large molecules. One of the practical merits of a monolith column is that the back pressure is lower than that of a particulate silica column27. Despite the high flow rate (400 µl/min) and low column temperature (20 °C), maximum back pressure of the system was 12.5 MPa37. This is the first demonstration of the advantage of a C18-bonded silica monolith column for the rapid analysis of a long oligonucleotide. Owing to the high flow rate, a dedicated instrument for micro LC and precise alignment at the interface are not required. Instead, a heated ESI probe is required to dissociate the DNA duplex and assist ionization of DNA as described later.
Ion-pair chromatography is commonly used for the MS-compatible separation of oligonucleotides. However, an ion-pair reagent generally interferes with the ESI process and decreases sensitivity of ESI-MS. Therefore, HFIP is frequently used for the mobile phase to improve the sensitivity of the oligonucleotide. However, HFIP (boiling point 59 °C) vaporizes rapidly at the interface before methanol (boiling point 65 °C) and, therefore, this loss of solvent increases pH and promotes dissociation of the ion-pair reagent (i.e., TEA) from the oligonucleotide. Because the present method employs a heated ESI probe, which nebulizes the eluate with hot nitrogen gas at 350 °C, this effect may be over-emphasized. Instead of HFIP-TEA buffer, Erb and Oberacher recommended cyclohexyldimethylammonium acetate (CycHDMAA; pH 8.4) for genotyping analysis because of a reduction in adduct formation with trace metal ions33. The authors deduced that CycHDMAA itself suppressed the formation of the metal adduct. Despite the literature, significant adduct formation has not been observed in the present method. Additionally, the noteworthy benefit of the HFIP-TEA-methanol system is that the peak area obtained with the HFIP-TEA-methanol system was 17 times greater than that obtained from the CycHDMAA-acetonitrile system when analyzing an 86 bp amplicon of T. poecilonotus (data not shown). One disadvantage of the HFIP-TEA-methanol system, however, is the increased cost relative to the CycHDMAA-acetonitrile system.
Calculation of the monoisotopic mass requires the separation of isotopic peaks of multiply charged ions. Therefore, resolution is critical for the present analysis. Although requisite resolution power is dependent on the base pair length of the analyte, conventional TOF analyzers, which have a resolution power of several tens of thousands, may be limited for the analysis of short oligonucleotides.
The theoretical monoisotopic masses shown in Table 4 were calculated from the corresponding base compositions of the analytes. Alternatively, Muddiman et al. developed a software application to calculate the base composition from the accurate mass43. A similar program was integrated into the automated ESI-MS system16-18. The use of these algorithms may improve the robustness of the present method because a measured monoisotopic mass does not always correspond to a unique base composition owing to the mass tolerance of 3 ppm resulting from the inevitable measurement error. Unfortunately, we could not obtain these software products for the present study.
The present monoisotopic mass-based species determination may be suitable not only for the differentiation of pufferfish but also for the detection of other DNA polymorphisms because the present method is based on analyzing the base composition and does not involve a procedure specifically designed for pufferfish. As for the detection of DNA polymorphism, the dedicated ESI-MS system is fully automated and easy to operate, which may be suitable for diagnostic use such as detection of pathogens15,17,18,20,30. Conversely, the present method is feasible with common research instruments and apparatus and, therefore, suitable for research uses. ESI-MS has already been applied to human DNA polymorphism such as single nucleotide polymorphism13,28,32, short tandem repetition26, and mitochondrial DNA analsis16,19. Micro RNA was also analyzed via capillary LC/ESI-MS44. These published applications can also be realized by the present method. In addition, this method may be suitable for monitoring the interaction between oligonucleotide and low-molecular-weight compounds, such as the interaction of antibiotics and ribosomal RNA45 owing to the low-temperature separation. In such cases, oligonucleotides and low-molecular-weight compounds should be detected simultaneously, which is an advantage of using LC/ESI-MS.
There is a limitation that the instrumentation is not suitable for performing parallel analysis as in conventional techniques such as Sanger sequencing and real-time PCR. Additionally, the present method identifies only base composition and any base substitution within the same molecule cannot be distinguished. However, the MS-based DNA analysis described here may still have merit in terms of accuracy in comparison with the DNA binding dye-based techniques such as gel electrophoresis and real-time PCR.
Divulgaciones
The authors have nothing to disclose.
Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research by the Japanese Society for the Promotion of Science (15K08060).
Materials
| DNeasy Blood & Tissue Kit (50) | Qiagen | 69504 | DNA extraction kit |
| Proteinase K | Qiagen | 19131 | |
| Ethanol (99.5+ vol%) | Wako | 054-07225 | For dilution of wash buffers |
| TE buffer (pH 8.0) | Wako | 310-90023 | For dilution of DNA sample |
| PCR primer | Fasmac | NA | Purified with reverse-phase cartridge column by the supplier |
| Template DNA | Eurofins Genomics | NA | Purified by HPLC by the supplier |
| Ultrapurified water | NA | NA | Generated with a Milli-Q Direct water purification system (Merck Millipore), used for sample preparation |
| Detergent Free 5X Phusion HF Buffer | Thermo Fisher | F-520L | Use instead of the provided buffer of DNA polymerase |
| Pfu-X DNA polymerase | Jena Bioscience | PCR-207S | |
| dNTP mix (20 mM each) | Jena Bioscience | NA | Supplied with the DNA polymerase |
| 10× Universal buffer M | Takara Bio | NA | Containing 100 mM Tris-HCl (pH 7.5), 100 mM MgCl2, 10 mM dithiothreitol and 500 mM NaCl |
| Dra I | Thermo Fisher | FD0224 | Restriction enzyme |
| Msp I | Thermo Fisher | FD0544 | Restriction enzyme |
| 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) | Fluka | 42060-50ML | Eluent additive for LC-MS grade. |
| Triethylamine (TEA) | Fluka | 65897-50ML | Eluent additive for LC-MS grade. |
| Trifluoroacetic acid | Sigma-Aldrich | T6508 | For preparation of calibrant. |
| Sodium hydroxide (1.0 M) | Fluka | 72082 | Dilute to 10 mM with ultrapurified water and use for titration of trifluoroacetic acid. |
| Acetonitrile | Fluka | 34967 | For preparation of calibrant, LC/MS grade. |
| Methanol | Kanto | 25185-76 | For mobile phase, LC/MS grade. |
| Water | Thermo Fisher | W6-1 | For mobile phase, LC/MS grade. |
| Microcentrifuge tube (0.5 ml) | Eppendorf | 0030123301 | PCR clean grade |
| Microcentrifuge tube (1.5 ml) | Eppendorf | 0030123328 | PCR clean grade |
| UVette | Eppendorf | 0030106300 | Disposable UV cuvette. |
| Gel Green | Biotium | 41004 | Fluorescent DNA stain |
| Cosmospin filter G (0.2 μm) | Nakalai Tesque | 06549-44 | Made of hydrophilic polytetrafluoroethylene (PTFE) membrane. Any centrifugal filter unit (pore size, 0.2–0.5 μm) made of hydrophilic PTFE or another low-binding membrane is applicable. |
| 300-μl PP screw vial | American Chromatography Supplies | V0309P-1232 | |
| Preassembled screw cap and septa | Finneran | 5395-09R | |
| Monobis C18 analytical column (2.0×50 mm, mesopore size = 30 nm) | Kyoto Monotech | 2050H30ODS | Outside Japan, available for purchase from GL Sciences via its distributors. (http://www.glsciences.com/distributors/) |
| Monobis C18 guard column | Kyoto Monotech | GCSET-ODS3210 | Holder included. |
| Cadenza CW-C18 (3.0×250 mm) | Imtakt | CW036 | C18-bonded particulate silica column |
| ProSwift RP-4H (1.0×250 mm) | Thermo Fisher | 066640 | Poly(styrene-divinylbenzene) monolith column |
| Themo Mixer C | Eppendorf | 5382 000.023 | For digestion of fish tissues. |
| Spectrophotometer | Shimadzu | UV-3150 | Quantification of DNA concentration. |
| Thermal cycler | Bio-Rad | T100 | |
| Portable refrigerator | Twinbird | D-CUBE | For aqueous mobile phase to avoid evaporation of HFIP. This can be replaced by an ice box. |
| Ultimate 3000 liquid chromatograph | Thermo Fisher | NA | |
| Q Exactive mass spectrometer | Thermo Fisher | NA | Fourier transform mass spectrometer equipped with the modified Kingdon trap analyzer. |
| Protein deconvolution 3.0 | Thermo Fisher | NA | Use version 3.0 or higher having an isotopic patter model of nucleotide. |
Referencias
- Oberacher, H. On the use of different mass spectrometric techniques for characterization of sequence variability in genomic DNA. Anal. Bioanal. Chem. 391, 135-149 (2008).
- Banoub, J. H., Miller-Banoub, J., Jahouh, F., Joly, N., Martin, P., Banoub, J. H., Limbach, P. A. Chapter 1, Overview of recent developments in the mass spectrometry of nucleic acid and constituents. Mass spectrometry of nucleosides and nucleic acids. 1, 1-90 (2010).
- Yergey, J., Heller, D., Hansen, G., Cotter, R. J., Fenselau, C. Isotopic distributions in mass spectra of large molecules. Anal. Chem. 55, 353-356 (1983).
- Zubarev, R. A., Demirev, P. A. Isotope depletion of large biomolecules: Implications for molecular mass measurements. J. Am. Soc. Mass Spectrom. 9, 149-156 (1998).
- Null, A. P., Muddiman, D. C. Determination of a correction to improve mass measurement accuracy of isotopically unresolved polymerase chain reaction amplicons by electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry. Rapid Commun. Mass Spectrom. 17, 1714-1722 (2003).
- Hu, Q., Noll, R. J., Li, H., Makarov, A., Hardman, M., Cooks, R. G. The Orbitrap: a new mass spectrometer. J Mass Spectrom. 40, 430-443 (2005).
- Senko, M. W., Beu, S. C., McLaffertycor, F. W. Determination of monoisotopic masses and ion populations for large biomolecules from resolved isotopic distributions. J. Am. Soc. Mass Spectrom. 6, 229-233 (1995).
- Horn, D. M., Zubarev, R. A., McLafferty, F. W. Automated reduction and interpretation of high resolution electrospray mass spectra of large molecules. J. Am. Soc. Mass Spectrom. 11, 320-332 (2000).
- Frahm, J. L., Mason, C. J., Muddiman, D. C. Utility of accurate monoisotopic mass measurements to confidently identify lambda exonuclease generated single-stranded amplicons containing 7-deaza analogs by electrospray ionization FT-ICR mass spectrometry. Int. J. Mass Spectrom. 234, 79-87 (2004).
- Frahm, J. L., Muddiman, D. C. Nucleic Acid analysis by fourier transform ion cyclotron resonance mass spectrometry at the beginning of the twenty-first century. Curr. Pharm. Des. 11, 2593-2613 (2005).
- Null, A. P., George, L. T., Muddiman, D. C. Evaluation of sample preparation techniques for mass measurements of PCR products using ESI-FT-ICR mass spectrometry. J. Am. Soc. Mass Spectrom. 13, 338-344 (2002).
- Null, A. P., Benson, L. M., Muddiman, D. C. Enzymatic strategies for the characterization of nucleic acids by electrospray ionization mass spectrometry. Rapid Commun. Mass Spectrom. 17, 2699-2706 (2003).
- Manduzio, H., Ezan, E., Fenaille, F. Evaluation of the LTQ-Orbitrap mass spectrometer for the analysis of polymerase chain reaction products. Rapid Commun. Mass Spectrom. 24, 3501-3509 (2010).
- Manduzio, H., Martelet, A., Ezan, E., Fenaille, F. Comparison of approaches for purifying and desalting polymerase chain reaction products prior to electrospray ionization mass spectrometry. Anal. Biochem. 398, 272-274 (2010).
- Hofstadler, S. A., et al. TIGER: the universal biosensor. Int. J. Mass Spectrom. 242, 23-41 (2005).
- Eduardoff, M., et al. Mass spectrometric base composition profiling: Implications for forensic mtDNA databasing. Forensic Sci. Int. Genet. 7, 587-592 (2013).
- Tang, Y. W., et al. Clinical accuracy of a PLEX-ID flu device for simultaneous detection and identification of influenza viruses A and B. J. Clin. Microbiol. 51, 40-45 (2013).
- Legoff, J., et al. Broad-range PCR-electrospray ionization mass spectrometry for detection and typing of adenovirus and other opportunistic viruses in stem cell transplant patients. J. Clin. Microbiol. 51, 4186-4192 (2013).
- Kiesler, K. M., Coble, M. D., Hall, T. A., Vallone, P. M. Comparison of base composition analysis and Sanger sequencing of mitochondrial DNA for four U.S. population groups. Forensic Sci. Int. Genet. 8, 226-232 (2014).
- Bacconi, A., et al. Improved sensitivity for molecular detection of bacterial and Candida infections in blood. J. Clin. Microbiol. 52, 3164-3174 (2014).
- Huber, C. G., Oberacher, H. Analysis of nucleic acids by on-line liquid chromatography-mass spectrometry. Mass Spectrom. Rev. 20, 310-343 (2001).
- Pourshahian, S., McCarthy, S. M., Bonilla, J. V., Srivatsa, G. S. Chapter 4, Analysis of oligonucleotides by liquid chromatography and mass spectrometry. Handbook of Analysis of Oligonucleotides and Related Products. , 137-166 (2011).
- Apffel, A., Chakel, J. A., Fischer, S., Lichtenwalter, K., Hancock, W. S. Analysis of Oligonucleotides by HPLC-Electrospray Ionization Mass Spectrometry. Anal. Chem. 69, 1320-1325 (1997).
- Premstaller, A., Oberacher, H., Huber, C. G. High-performance liquid chromatography-electrospray ionization mass spectrometry of single- and double-stranded nucleic acids using monolithic capillary columns. Anal. Chem. 72, 4386-4393 (2000).
- Oberacher, H., Oefner, P. J., Parson, W., Huber, C. G. On-Line Liquid Chromatography Mass Spectrometry: A Useful Tool for the Detection of DNA Sequence Variation. Angew. Chem. Int. Ed. Engl. 40, 3828-3830 (2001).
- Oberacher, H., Parson, W., Muhlmann, R., Huber, C. G. Analysis of polymerase chain reaction products by on-line liquid chromatography-mass spectrometry for genotyping of polymorphic short tandem repeat loci. Anal. Chem. 73, 5109-5115 (2001).
- Oberacher, H., Huber, C. G. Capillary monoliths for the analysis of nucleic acids by high-performance liquid chromatography-electrospray ionization mass spectrometry. Trends Anal. Chem. 21, 166-174 (2002).
- Oberacher, H., et al. Re-sequencing of multiple single nucleotide polymorphisms by liquid chromatography-electrospray ionization mass spectrometry. Nucleic Acids Res. 30, e67 (2002).
- Oberacher, H., Parson, W., Holzl, G., Oefner, P. J., Huber, C. G. Optimized suppression of adducts in polymerase chain reaction products for semi-quantitative SNP genotyping by liquid chromatography-mass spectrometry. J. Am. Soc. Mass Spectrom. 15, 1897-1906 (2004).
- Mayr, B. M., et al. Identification of bacteria by polymerase chain reaction followed by liquid chromatography-mass spectrometry. Anal. Chem. 77, 4563-4570 (2005).
- Oberacher, H., Niederstatter, H., Casetta, B., Parson, W. Some guidelines for the analysis of genomic DNA by PCR-LC-ESI-MS. J. Am. Soc. Mass Spectrom. 17, 124-129 (2006).
- Beer, B., et al. CYP2D6 genotyping by liquid chromatography-electrospray ionization mass spectrometry. Anal. Bioanal. Chem. 400, 2361-2370 (2011).
- Erb, R., Oberacher, H. Comparison of mobile-phase systems commonly applied in liquid chromatography-mass spectrometry of nucleic acids. Electrophoresis. 35, 1226-1235 (2014).
- Cohen, N. J., et al. Public health response to puffer fish (Tetrodotoxin) poisoning from mislabeled product. J. Food Prot. 72, 810-817 (2009).
- Cole, J. B., et al. Tetrodotoxin poisoning outbreak from imported dried puffer fish–Minneapolis, Minnesota 2014. MMWR Morb. Mortal. Wkly. Rep. 63, 1222-1225 (2015).
- Ohno, Y., et al. The influence of tetrodotoxin on the toxic effects of aconitine in vivo. Tohoku J. Exp. Med. 167, 155-158 (1992).
- Miyaguchi, H., Yamamuro, T., Ohta, H., Nakahara, H., Suzuki, S. Genotyping of Toxic Pufferfish Based on Specific PCR-RFLP Products As Determined by Liquid Chromatography/Quadrupole-Orbitrap Hybrid Mass Spectrometry. J. Agric. Food Chem. 63, 9363-9371 (2015).
- Sambrook, J. F., Russell, D. W. . Neutral polyacrylamide gel electrophoresis. in: Molecular Cloning: A Laboratory Manual. 1, 40-46 (2001).
- Moini, M., Jones, B. L., Rogers, R. M., Jiang, L. Sodium trifluoroacetate as a tune/calibration compound for positive- and negative-ion electrospray ionization mass spectrometry in the mass range of 100-4000 Da. J. Am. Soc. Mass Spectrom. 9, 977-980 (1998).
- Fountain, K. J., Gilar, M., Gebler, J. C. Analysis of native and chemically modified oligonucleotides by tandem ion-pair reversed-phase high-performance liquid chromatography/electrospray ionization mass spectrometry. Rapid Commun. Mass Spectrom. 17, 646-653 (2003).
- Chen, B., Bartlett, M. G. Evaluation of mobile phase composition for enhancing sensitivity of targeted quantification of oligonucleotides using ultra-high performance liquid chromatography and mass spectrometry: application to phosphorothioate deoxyribonucleic acid. J. Chromatogr. A. 1288, 73-81 (2013).
- Gong, L., McCullagh, J. S. Analysis of oligonucleotides by hydrophilic interaction liquid chromatography coupled to negative ion electrospray ionization mass spectrometry. J. Chromatogr. A. 1218, 5480-5486 (2011).
- Muddiman, D. C., Anderson, G. A., Hofstadler, S. A., Smith, R. D. Length and base composition of PCR-amplified nucleic acids using mass measurements from electrospray ionization mass spectrometry. Anal. Chem. 69, 1543-1549 (1997).
- Kullolli, M., Knorf, E., Arampatzidou, M., Tewari, M., Pitteri, S. J. Intact MicroRNA analysis using high resolution mass spectrometry. J. Am. Soc. Mass Spectrom. 25, 80-87 (2013).
- Cummins, L. L., Chen, S., Blyn, L. B., Sannes-Lowery, K. A., Drader, J. J., Griffey, R. H., Hofstadler, S. A. Multitarget affinity/specificity screening of natural products: finding and characterizing high-affinity ligands from complex mixtures by using high-performance mass spectrometry. J. Nat. Prod. 66, 1186-1190 (2003).
