Quality of DNA prepared using the combined HotShot/Drop-Dialysis is sufficient for satisfactory digital PCR (Figure 1B). For 2,000 fibroblasts, approximately 3,000 ± 500 positive droplets were obtained, corresponding to 75% of the expected 4,000 copies (two copies in one cell). In this study, the tumor cells were from an established line MPNST S462 which are known to have a heterozygotic deletion of the NF1 gene5. This 50% gene dosage reduction was clearly detected by the dual digital PCR assay (Figure 1B). By contrast, two copies of this gene were measured in fibroblasts. This genetic difference between tumor and non-tumor cells enables calculation of the proportion of tumor cells in mixed cultures (Figure 2). By measuring the relative gene dosage of NF1 a RPP30, proportion of tumor cells in a mixed culture can be determined at each drug-concentration (Figure 3).
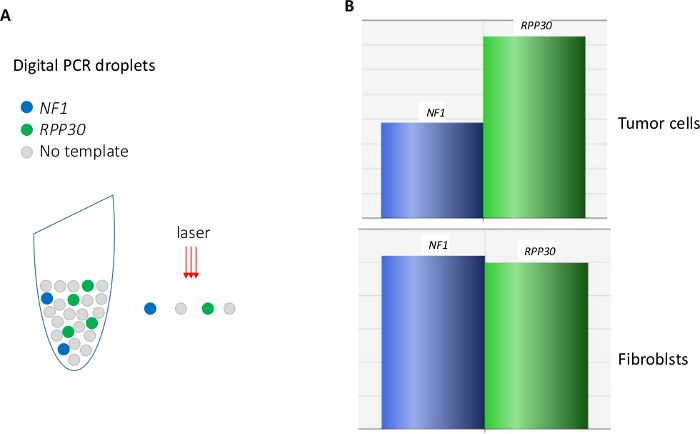
Figure 1. Digital PCR. (A) illustration of the principle. (B) a dual-probe assay reveals reduced amount of a target (NF1) gene in the tumor cells MPNST S462 in relation to a reference (RPP30) gene. Please click here to view a larger version of this figure.
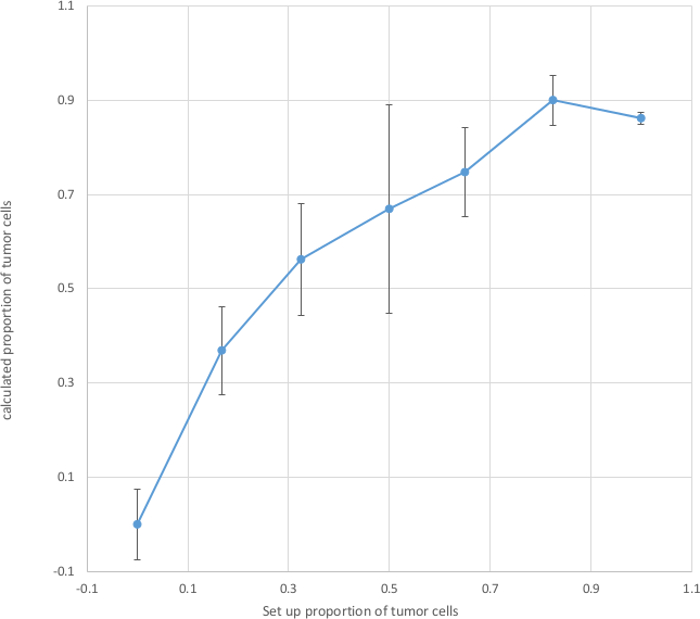
Figure 2. Calculated Proportion of Tumor Cells. Tumor cells and non-tumor cells were mixed at various ratios and seeded into each well of a 96-plate. After attachment O/N, the cells were lysed with HotShot/Drop-dialysis and the desalted lysates were subjected to digital PCR which gave the ratio of NF1/RPP30. Base on this ratio, proportion of tumor cells in each well was calculated. Mean and standard deviation of proportion of tumor cells were calculated from 4 replicates at each set-up proportion. The calculated proportions of tumor cells (Y-axis) were plotted against the set-up proportions (X-axis). Please click here to view a larger version of this figure.
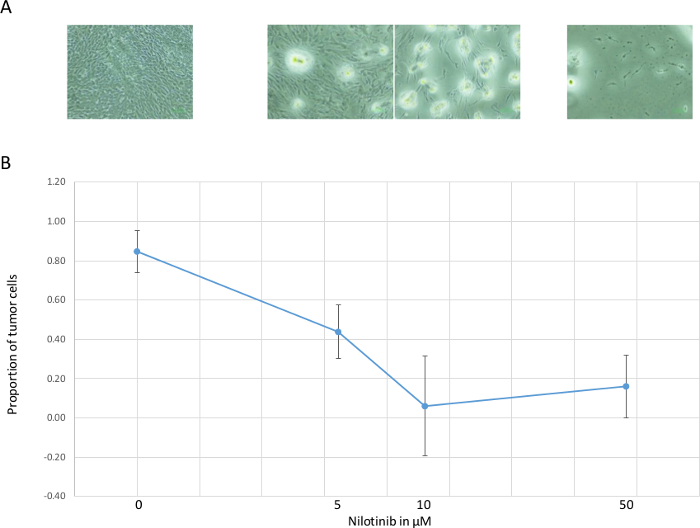
Figure 3. Dose-dependent Change of Proportion of Tumor Cells. (A) dose-dependent reduction of visible vital cells. (B) proportion of tumor cells (mean and standard deviation of 4 replicates) calculated from the ratio of NF1: RPP30, decreased in the range of 0 – 10 µM. At the highest dose 50 µM, few vital cells were left, likely due to toxic effect to all cells. Please click here to view a larger version of this figure.
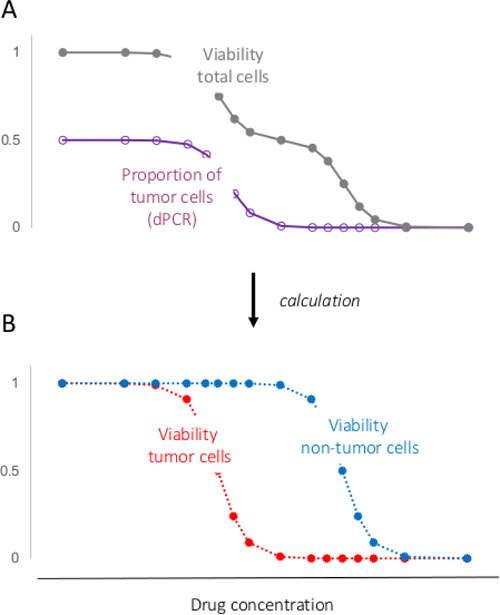
Figure 4. Separating Response of Tumor Cells from That of Non-tumor Cells. (A) viability or/and proliferation of total cells can be measured using conventional assays; Proportion of tumor cells can be determined using the procedure demonstrated in the present study. (B) using the two parameters, response of total cells in a mixed culture to a drug can be divided into response of the tumor cells and response of the non-tumor cells. Please click here to view a larger version of this figure.
| 1 Reaction | ||
| 2 x ddPCR supermix | 12 | |
| Assay mix for NF1 | 1.2 | |
| Assay mix for RPP30 | 1.2 | |
| HaeIII | 0.6 | |
| Subtotal | 15 | Master mixture |
| DNA | 9 | |
| Total | 24 | Final PCR Mixture |
Table 1 Setting Up Dual ddPCR Reaction
| Step | Action | Temperature | Time | Cycles |
| 1 | Polymerase activation | 95 °C | 10 min | 1 |
| 2 | DNA denaturation | 94 °C | 30 sec | 40 |
| 3 | Annealing/exetension | 60 °C | 1 min | |
| 4 | Polymerase deactivation | 98 °C | 10 min | 1 |
| 5 | Hold (optional) | 4 °C |
Table 2 PCR Cycler Settings
