In the first step of the protocol, the goal is to encapsulate the monometallic or bimetallic transition metal oxide (TMO) NPs within microporous silica spheres. Figure 1 shows images taken of representative syntheses before and after precipitation with methanol. Two reproducible morphological outcomes have been observed during this step that appear to be dependent on the metals used in the synthesis: the TMO NPs can be singly coated with a silica sphere (Figure 2b) or multiple TMO NPs can be coated within a single silica sphere (Figure 2a). Singly-coated TMO NPs have been observed for syntheses using Ti, Ta, and Nb, while multiply-coated TMO NPs have been observed for Mo and W. It is possible to synthesize these metals also in singly-coated formulations by performing the metal alkoxide hydrolysis at elevated pH. Table 1 details results for controlling particle size and composition using the RME. We hypothesize that at elevated pH, the rate of NP growth is higher, and larger NPs can more easily nucleate SiO2 sphere growth (Figure 6c, d). At low pH, certain metal alkoxides hydrolyze very slowly, resulting in ultrasmall TMO nuclei that ultimately become embedded in SiO2 spheres (Figure 6a, b).
The metal composition of the TMO NPs is controlled by the metal alkoxides added to the RME and whether they are mixed together (as presented in the given protocol) or added sequentially. For instance, TaIPO could be mixed with WIPO and heptane before injecting into the RME, or a TaIPO/heptane mixture could be injected into the RME 4 hr after a WIPO/heptane mixture has been injected and allowed to hydrolyze.
Many variables must be considered to control the size of the TMO NPs. The first set of variables is the selection of the surfactant and the oil phase. Here, the water/n-heptane/Brij-L4® system has been chosen due to its wide stability window and the ability to synthesize very small nanoparticles. Alternatively, water/n-heptane/Igepal CO-520® can be used if larger TMO NPs are desired as shown in Figure 3. Alternatively, one can modify the rates of nucleation vs. growth by adding NH4OH to the RME before metal alkoxide addition, resulting in larger NPs as shown in Figure 6c, and d, and outlined in Table 1. Once the RME system is chosen, the first set of variables controlling TMO size can be grouped together as RME control parameters. These include the water:surfactant ratio, the oil:water ratio, the oil:surfactant ratio, the temperature of the RME, and the extent of convective mixing. These parameters dictate the size of the suspended water droplets, their proximity to one another within the emulsion, their average polydispersity, and the rates of droplet coalescence and separation.
The final set of variables affecting TMO NP size can be classified as metal alkoxide hydrolysis control parameters. These include the metal alkoxide:water molar ratio, the length of time the metal alkoxide is allowed to hydrolyze before initiating the silica coating, the pH of the water droplets, the temperature, and the rate of metal alkoxide addition to the RME system.
Ultimately, the goal of this method is to produce non-sintered and metal-terminated TMC and TMN nanoparticles (referred to as TMCN NPs for convenience). Obtaining this result hinges on synthesizing microporous silica coatings with high thermal stability that also inhibit the sintering of TMC and TMN nanoparticles. To achieve this result, there are two sets of variables that must be considered: control variables affecting the thermal stability of the silica, and control variables affecting the TMCN particle size distribution (PSD).
On heating, microporous silica can transition to dense phases and ultimately to quartz, making it difficult to obtain phase-pure TMCN NPs and can make silica removal more challenging. To maximize the thermal stability of the silica coatings, it has been observed that a high pH is required during TEOS hydrolysis and that the SiO2/TMO NPs should be precipitated from the RME using methanol as opposed to other common precipitating agents such as acetone or isopropanol. Figure 4 shows powder x-ray diffractograms (PXRD) of carburized SiO2/WOx materials with the silica coatings performed at different pH values while Figure 5 shows PXRD diffractograms of carburized SiO2/WOx materials with the silica coatings performed at the same pH but precipitated with three different solvents. TEOS hydrolysis at high pH has been shown to lead to a high number of undercoordinated Q2 and Q3 sites, resulting in a higher micropore volume.28 The influence of the precipitating agent on silica thermal stability is poorly understood but TEM images suggest that flocculation with methanol leads to less aggregated SiO2/TMO flocs as compared with acetone and isopropanol (results not shown). We find support for this hypothesis from TEM images of SiO2/TMO flocs precipitated with less than the recommended amount of methanol in the procedure. For syntheses flocculated with less methanol, the flocs appear more aggregated and the silica is less stable, transitioning to quartz-like domains at lower temperatures than when excess methanol is used during the flocculation step (results not shown).
The TMCN PSD is controlled by the size of the initial TMO NPs as well as the silica nanostructure. In theory, if TMCN sintering can be fully mitigated by the silica shells at carburizing/nitridizing conditions, then the TMCN PSD will be fully controlled by the initial TMO PSD, adjusted by the density difference between the initial hydrated, amorphous TMO lattice and the final crystalline TMC or TMN lattice. Such a result has been closely achieved by using thick silica shells with representative examples shown in Figure 6c, and d.
If sintering is not fully mitigated, then the final TMCN PSD will be controlled by both the initial TMO PSD and the silica coating. This is particularly true for thin silica coatings or for ultrasmall 1-2 nm TMO NPs that can more easily diffuse within the silica coatings. A representative example is shown in Figure 6a and 6b. Here, the same initial 1-2 nm TMO PSD is used, but the silica coating is altered from 50 nm to 35 nm. In the thick silica coating, sintering is mitigated and a 1-2 nm TMC PSD is obtained while in the thin silica coating, sintering is only partially mitigated and a 2-3 nm TMC PSD results. Note that some sintering is present at the surface of silica spheres post-carburization, which we attribute to small surface-bound TMC NPs that can laterally diffuse across the surface of the silica spheres and sinter.
After carburization, it has been observed that for multiply-coated TMCN NPs, the silica coatings remain spherical with minimal sintering of the silica (Figure 6a and 6b). In contrast, for singly-coated TMCN NPs, the silica coatings sinter together (Figure 6c and 6d). We hypothesize that multiply-coated TMCN NPs provide structural integrity to the silica spheres at high temperatures, preventing the sintering of the silica spheres. While this is not the case for singly-coated TMCN NPs, the sintering of the silica spheres has not been observed to inhibit the ability of the silica coatings to both prevent sintering of the TMCN NPs while also allowing carburizing or nitridizing gas molecules to diffuse through them (Figure 6c and 6d). PXRD diffractograms have been included in Figure 8 for various monometallic and heterometallic early transition metal carbide and nitride nanoparticles of various sizes.
Dissolution of the TMCN NPs onto carbon black (CB) leads such as Vulcan XC-72r to well-dispersed, supported NPs. A representative result is shown in Figure 7c. Alternatively, if no support is added, a black nanodispersion suspension is obtained as shown in Figure 7a. Because no surface stabilizing agents are added during or after the dissolution, the TMCN NPs form small aggregates in solution, a representative example of which is shown in Figure 7b.
| Experimental Result | Precursors | Volume (ml) | Initial NH4OH (ml) | Final NH4OH (ml) | TEOS (ml) |
| 1-2 nm WC | W(VI)IPO (5% w/v) | 12 | 0 | 2.7 | 1.2 |
| 2-3 nm WC | W(VI)IPO (5% w/v) | 12 | 0 | 1.4 | 0.6 |
| 6-8 nm WC | W(VI)IPO homemade (5% w/v) | 12 | 0.4 | 1.4 | 1.6 |
| 7-10 nm WC | W(IV)IPO homemade (6.5% w/v) | 8.8 | 1.4 | 1.4 | 1.6 |
| 9-13 nm WC | W(IV)IPO homemade (6.5% w/v) | 10.2 | 1.4 | 1.4 | 1.6 |
| 4-6 nm Ti0.1W0.9C | W(VI)IPO (5% w/v) | 10.8 | 0.7 | 1.4 | 0.6 |
| Ti(IV)IPO (5% w/v) | 0.7 | ||||
| 7-10 nm (Ni0.3W0.7)2C | W(VI)IPO (5% w/v) | 8.4 | 0.4 | 1.4 | 0.6 |
| Ni(II)MEO (5% w/v) | 1.4 |
Table 1: Controlling TMC particle size by controlling RME parameters. *Initial NH4OH is if NH4OH has been added to the emulsion before metal alkoxide hydrolysis while final NH4OH is the total amount of NH4OH present in the emulsion before TEOS is injected.

Figure 1: Representative pictures of RME syntheses containing SiO2/TMO NPs immediately before (A – C) and after (D, F) the addition of 300 ml of methanol. (A) shows a synthesis of SiO2/WOx, (B) shows an SiO2/Mo0.06W0.94Ox synthesis, and (C) shows an SiO2/Mo0.54W0.46Ox synthesis while (D) and (E) show different viewing angles of the SiO2/WOx synthesis after the addition of methanol. In (D) and (E), the top phase is the heptane-rich liquid phase, the lower liquid phase is methanol-rich, and at the bottom of the flask are the SiO2/WOx flocs. Re-print with permission from reference 26.

Figure 2: Representative TEM images of (A) multiply-coated WOx NPs within SiO2 spheres and (B) singly-coated Ta0.3W0.7Ox NPs within SiO2 spheres. The scale bars are in nanometers. Modified from reference 26.

Figure 3: Representative TEM images of SiO2/WOx synthesized under identical conditions described in the procedure except with 60 ml of Igepal® CO-520 instead of 54 ml of the Brij®-L4 surfactant. The scale bar is in nanometers. Modified from reference 26.
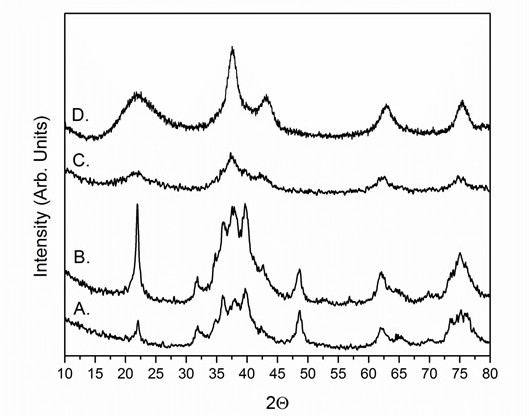
Figure 4: Representative PXRD diffractograms of SiO2/WOx coated at different pH values. Carburizations were conducted at 835 °C for 4 hr under 21% CH4/H2 and are shown for materials coated using a pH of (A) 10, (B) 10.5, (C) 10.9, and (D) 11.1. The low pH syntheses (A and B) have quartz-like silica (indicated by the sharp peak at 22°) and multiple carbide phases while the high pH syntheses (C and D) have a broad peak centered at 22°, indicative of amorphous silica, and single-phase face-centered-cubic WC NPs. Re-print with permission from reference 26.
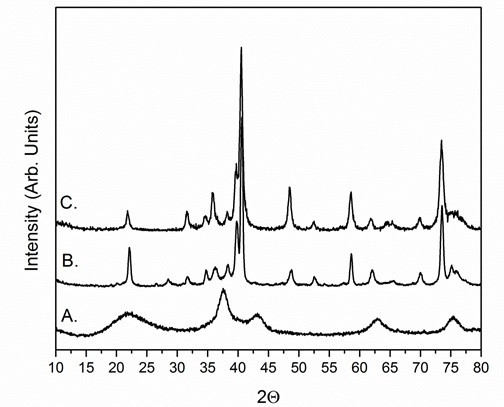
Figure 5: Solvent precipitation effect studies by precipitating SiO2/WOx or SiO2/MoxW1-xOy with (A) Methanol, (B) Acetone, and (C) Isopropanol. All materials were rinsed with acetone after precipitation. The PXRD diffractograms are for the resulting materials post-carburization at 835 °C for 4 hr under 21% CH4/H2. Re-print with permission from reference 26.
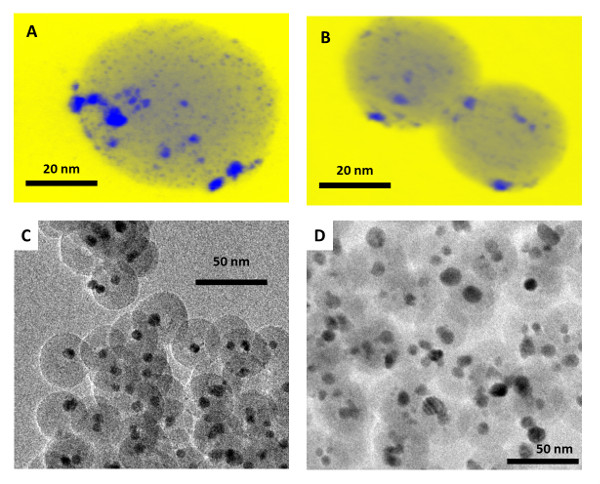
Figure 6: Representative TEM and HAADF-STEM images of (A) 1-2 nm SiO2/WC NPs post-carburization, (B) 2-3 nm SiO2/WC NPs post-carburization, (C) 7-10 nm SiO2/WC NPs post-carburization, (D) 9-13 nm SiO2/WC NPs post-carburization. The scale bars are in nanometers. Modified from reference 26.
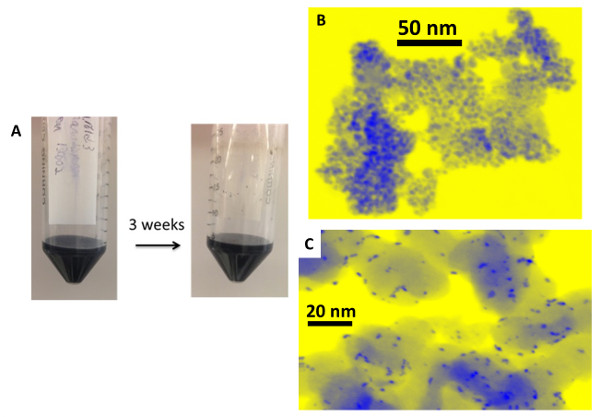
Figure 7: (A) photographs of a hexagonal WC nanodispersion dispersed in water at 7.5 mg/ml before and after three weeks of sitting in stagnant ambient conditions, (B) representative HAADF-STEM image of an unsupported β-WC nanopowder, (C) representative HAADF-STEM image of β-WC NPs supported on carbon black at 25 wt%. The scale bars are in nanometers. Modified from reference 26.
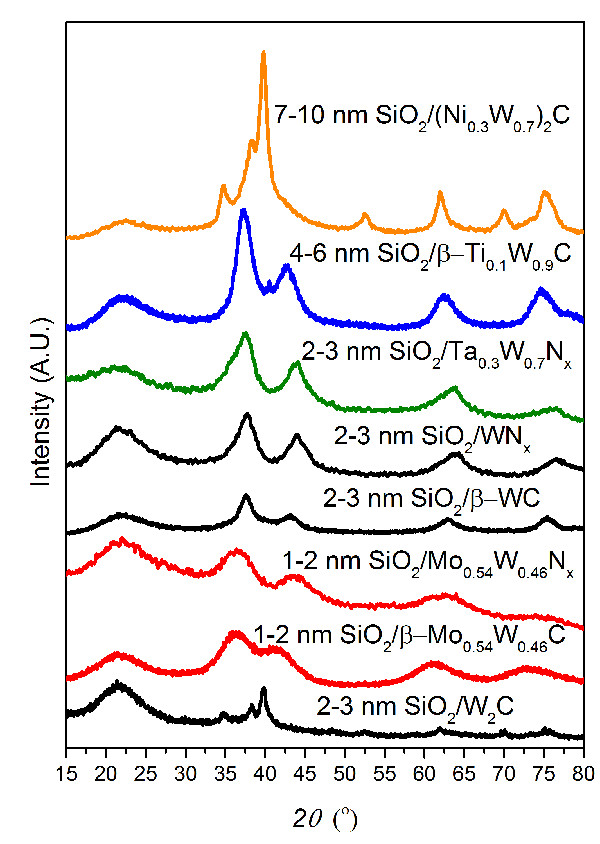
Figure 8: PXRD diffractograms of silica-encapsulated monometallic and heterometallic early transition metal carbides and nitrides of various sizes. All TMNs were nitridized under 100 sccm NH3 at 800 °C for 4 hr. 2-3 nm SiO2/W2C was synthesized at 775 °C under 18% CH4/H2 for 4 hr while all of the <3 nm TMC NPs were synthesized at 835 °C under 21% CH4/H2. The larger TMC NPs were synthesized at 900 °C under 21% CH4/H2. Modified from reference 26.
