Retaining the immunophenotype and functionality of microglia ex vivo during isolation is critical to be able to utilize these cells as an investigatory model for microglial biology. In order to demonstrate the successful preservation of microglia immunofunctionality using the present method, we isolated cortical microglia from P3 neonates (CX3CR1-GFP+/-and C57BL/6) and treated cultures with either LPS or Pam.
As illustrated in Figure 1A, isolation of microglia via agitation through rotary shaking preserves the quiescent phenotype attributed to microglia in normal in vivo conditions. To assess microglial purity, CX3CR1-GFP+/- cell cultures were stained for the macrophage antigen Iba-1 and counter stained with DAPI in order to visualize the cell nucleus. As shown in Figures 1A and 1B, under low magnification, microglia isolation via the presently described method results in a highly pure cell culture, as greater than 95% of cells exhibit colocalization of GFP, Iba-1, and DAPI.
We next sought to confirm the responsiveness of microglia ex vivo by challenging cells with LPS. In comparison to vehicle treatment, LPS-treated CX3CR1-GFP+/- microglia adapted a stark morphological change from a small, bipolar phenotype to an amoeboid-like shape, as shown in Figure 1B. This morphological change is also recapitulated in cortical microglia isolated from C57BL/6 neonates treated with Pam (Figure 2). This conserved morphological response between genetically distinct microglia, activated with different stimuli, underscores the reproducibility and applicability of the present protocol in regards to various experimental models.
Even though this change in morphology is indicative of activation39, it is necessary to determine if microglia isolated by rotary shaking retain their ability to translocate p65 upon activation, indicating the preservation of functional TLR-mediated intracellular signaling. To confirm this signaling cascade, we treated microglia isolated from C57BL/6 neonates with Pam. As illustrated in Figure 2, immunocytochemical staining for p65 evidenced that, under normal conditions, p65 is diffusely localized throughout the cytoplasm, whereas after a 2 hr stimulation with Pam, p65 immunoreactivity dramatically shifted localization to the cell nucleus.
Having established that microglia isolated via the method presented herein preserve the molecular mechanisms to translocate p65 to the nucleus, we next wanted to confirm that isolated microglia retain their biochemical immunofunctionality by testing their ability to secrete a hallmark proinflammatory cytokine, TNF-α, upon activation. As demonstrated in Figure 3, in comparison to vehicle-treated microglia, cells exposed to Pam or LPS increase the secretion of TNF-α into the culture media (P<0.0001; one-way ANOVA), indicative of functional TLR1/2 andTLR4 signaling pathways, respectively.
Therefore taken together, these data establish that isolation of microglia via rotary shaking results in highly pure cell cultures with preserved immunophenotype and functionality, ex vivo.
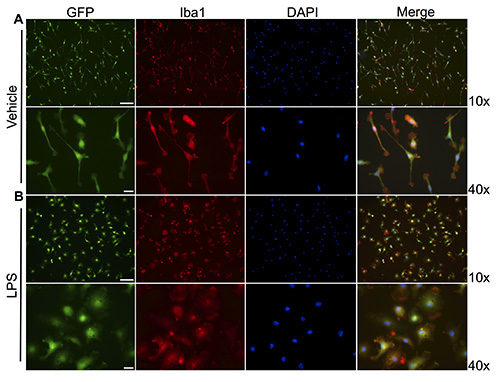
Figure 1. Rotary shaking yields high purity microglial cultures with preserved immunophenotype. Microglial cells were isolated from CX3CR1-GFP+/- neonatal mice at P3 and treated with either vehicle (A), or bacterial LPS (B) for 24 hr. Cells were fixed with 4% PFA, immunocytochemically stained for Iba-1, and counter stained with DAPI. (A) Microglia treated with PBS exhibit a bipolar, quiescent phenotype. (B) Upon LPS challenge, microglia adapt an amoeboid-like phenotype, indicative of activation. Merged channel in panels (A) and (B) under 10X magnification demonstrate that >95% of cells are GFP/Iba-1 positive cells. 10X scale bar: 100 μm; 40X scale bar: 20 μm. Click here to view larger image.
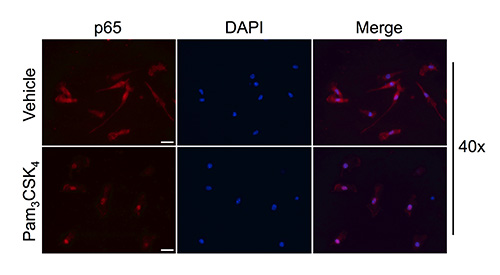
Figure 2. Microglia isolated via rotary shaking translocate NF-κB to cell nucleus upon Pam challenge. Microglial cells were isolated from C57BL/6 neonates at P3 and treated with vehicle or Pam for 2 hr. Cells were fixed with 4% PFA and stained for p65 via immunocytochemistry. Cells were counter stained with DAPI to visualize the cell nucleus. In vehicle treated microglia, p65 is localized throughout the cytoplasm, whereas stimulation with Pam induces the nuclear translocation of p65. 40X scale bar: 20 μm. Click here to view larger image.
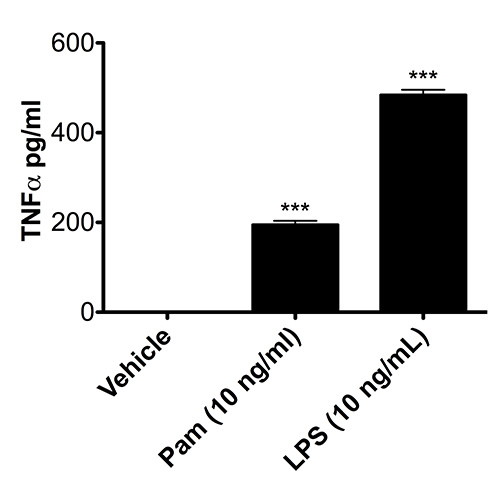
Figure 3. Microglia isolated with rotary shaking release TNF-α upon exposure to Pam or LPS. Microglial cells were isolated from C57BL/6 neonatal mice at P3 and treated with vehicle, Pam, or LPS for 2 hr. Cell culture supernatants were collected and analyzed via ELISA for TNF-α release. *** P< 0.0001 (n=2; one-way ANOVA). Data are presented as means ± SD.
