To visualize the interactions between mitochondria and lysosomes using this dual-color CLEM protocol, we employed Mito-mEosEM and TMEM192-V5-APEX2. Fixed cells were first imaged using a light microscope, followed by EM sampling and imaging. The results of the correlated images are shown in Figure 3A,B and Figure 4A. Following the protocol described above, we checked whether we could observe the changes in the interaction between mitochondria and lysosomes under external stress.
Treatment of cells with BFA, a lysosomal V-ATPase inhibitor, inhibits autophagy, resulting in reduced mitochondrial quality and increased interactions between mitochondria and lysosomes27. In BFA-treated cells, we observed mitochondria trapped within lysosomes (Figure 3B), compared to DMSO-treated control cells (Figure 3A). Although it is difficult to distinguish fragmented electron-dense mitochondria and cellular compartments within lysosomes in electron micrographs alone, they can be easily distinguished in correlated images (Figure 3B1,B2). The mitochondrial disruption structure (Figure 3A1,A2; blue arrow) observed in the mitochondria of DMSO-treated cells is not related to lysosomes, and the structure in the fluorescence image that appears to be a lysosome trapped inside a large mitochondrion is a structure where the mitochondrion surrounds the lysosome (Figure 3A2; red arrow).
Next, we tested the effects of U18666A (2 µg/mL for 18 h), a well-known inhibitor of NPC1 that blocks the movement of cholesterol out of lysosomes28. Cells treated with U18666A showed increased direct interaction between mitochondria and lysosomes (Figure 4). In addition, the contacts between mitochondria and lysosomes in U18666A-treated samples were different from those in BFA-treated samples. In HeLa cells treated with BFA, the fragmented mitochondrion is completely trapped in the lysosome, a previously reported mitophagy morphology12,29. However, in the U18666A-treated group, some of the TMEM192-positive lysosomes appear engulfed by mitochondrion (Figure 4D,E)30,31.
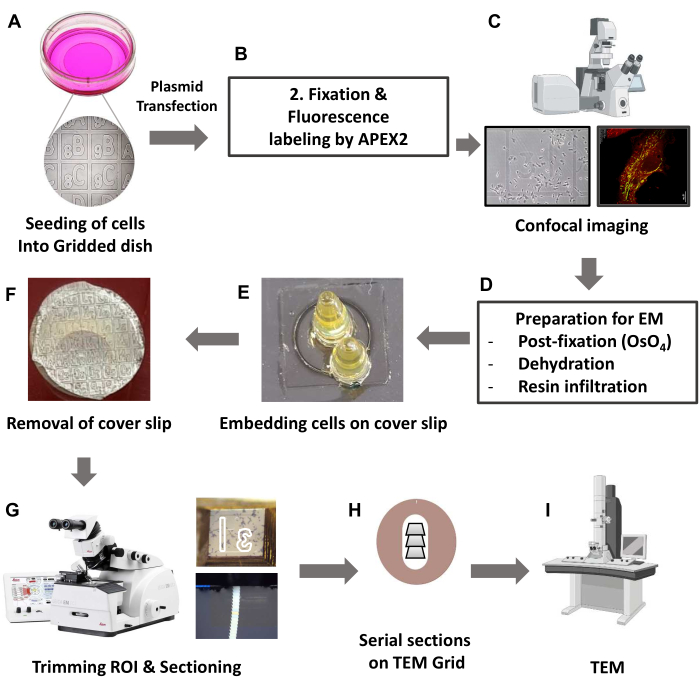
Figure 1: Schematic overview of TEM-based CLEM by using a genetically tagged fluorescence probe. (A) HeLa cells were grown in 35 mm glass grid-bottom culture dishes. (B) Fixed cells were labeled with Amplex-Red by APEX2 protein. (C) Fluorescence images were acquired with a confocal microscope. A motorized stage and tile-scan function were used to capture a large overview image and a high-resolution image of the target cell. The large image serves as a navigation map to identify the location of the target cell in plastic-embedded specimens. (D) Proceed with post fixation and dehydration embedding. (E) Embedding was done using a PCR tube or BEEM capsule filled with resin over the target cell on the coverslip. (F) After resin polymerization, removing the coverslip left an imprinted grid pattern on the bottom surface of the block. (G) The target cell was identified by the imprinted pattern on the resin surface, and the target cell was sectioned at approximately 60 nm thickness using an ultramicrotome. (H) Serial sections were collected on a formvar-coated copper slot grid. (I) The grids were imaged using transmission electron microscopy. Abbreviations: TEM = transmission electron microscopy; CLEM = correlative light and electron microscopy; ROI = region of interest. Please click here to view a larger version of this figure.
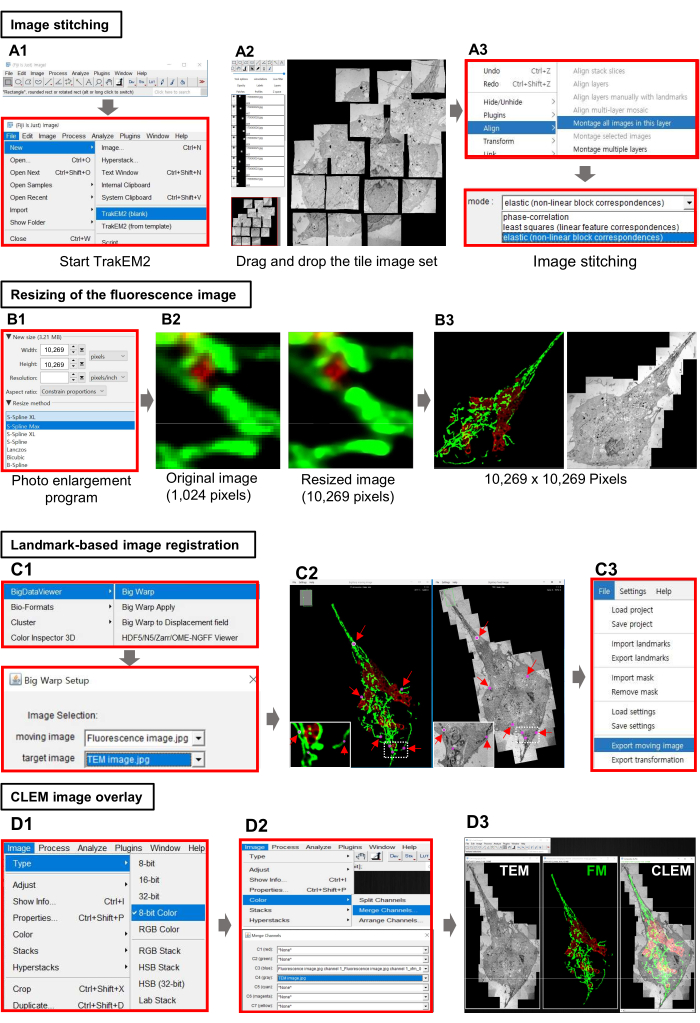
Figure 2: Image processing and generation of the CLEM image using imageJ Fiji. (A1) Main GUI of Fiji and TrakEM2. (A2) Import of the tile image dataset acquired with TEM at 1,700x magnification. (A3) Alignment and example of a stitched image using TrakeEM2's image alignment method. (B1–B3) Conversion of small light micrograph into large high-quality images using photo enlargement software. (C1–C3) Workflow for registration between light microscopy images and electron microscopy images using the Big Wrap plug-in software. (D1–D3) The process of overlaying a transformed FM image and an EM image to synthesize a correlated microscope image. Abbreviations: TEM = transmission electron microscopy; CLEM = correlative light and electron microscopy; FM = fluorescence microscopy. Please click here to view a larger version of this figure.

Figure 3: CLEM images of the mitochondrial and lysosomal contact sites treated with or without bafilomycin A1. HeLa cells were co-transfected with Mito-mEosEM (mitochondria; green) and TMEM192-APEX2 (lysosomes; red) and then stained with Amplex-Red dye. (A) Untreated HeLa cells. Molecular information from the fluorescence signal (left panel) and ultrastructural information obtained by TEM (middle panel) were superimposed (right panel) based on mitochondrial signal and morphology. (A1,A2) In untreated HeLa cells, the mitochondrial inner membrane is disrupted (blue arrow), or the mitochondria and lysosomes are very close to each other (red arrow). (B1,B2) In HeLa cells treated with bafilomycin A1 for 18 h, mitophagy, characterized by the trapping of undegraded mitochondria in lysosomes, was observed (green arrow). Scale bars = 1 µm (A1,A2,B1,B2). Please click here to view a larger version of this figure.
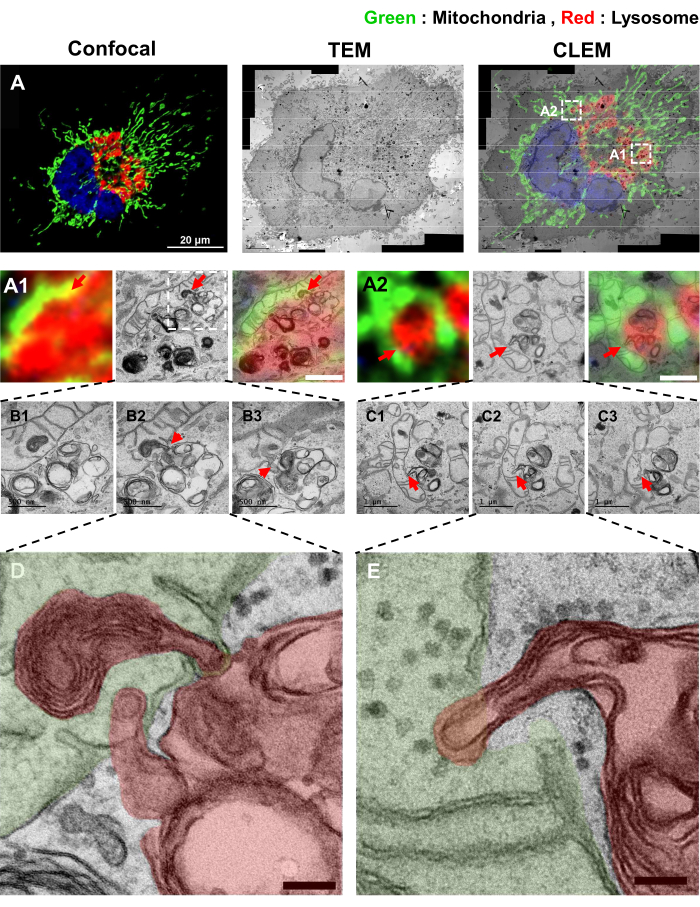
Figure 4: CLEM images of the mitochondrial and lysosomal contact sites treated with U18666A (A) Two-color CLEM images of Mito-mEosEM and TMEM192-V5-APEX2 (Amplex-Red-labeled) expressed HeLa cells under U18666A treatment (2 µg/mL for 18 h). (A1,A2) Red arrows indicate contact sites between mitochondria and lysosomes. Scale bars = 1 µm. (B1–B3,C1–C3) Red arrow indicates the appearance of inter-organelle fusion between mitochondria and lysosomes by serial images. Scale bars = 500 nm (B1–B3). Scale bars = 1 µm (C1–C3). (D,E) High-magnification TEM images of the fusion sites between mitochondria and lysosomes. Pseudocolors are used to distinguish between mitochondrion (green) and lysosome (red). Scale bars = 100 nm. Please click here to view a larger version of this figure.
