Using the setup described in step 3 (Figure 1A), nucleosome formation along the DNA tether was visualized as the appearance of red fluorescent foci on a 2D scan (Figures 1B, left) or trajectories over time on a kymograph (Figure 1B, right). Properly wrapped nucleosomes yielded fluorescence trajectories that were stationary over time within the diffraction limit of confocal detection (~300 nm). Notably, multiple nucleosomes formed near each other under the diffraction limit cannot be spatially resolved and appear as brighter foci or trajectories. Nevertheless, the photobleaching steps of individual nucleosomal trajectories were analyzed and frequently showed 1-step (Figure 1C) or 2-step (Figure 1D) photobleaching events, which indicates that foci mostly contained mononucleosomes given the approximately 70% labeling rate of the H4 protein in this sample. Moreover, based on the average photon count for a single LD655 fluorophore in this experimental condition, the number of labeled H4 copies in each nucleosome trajectory was estimated and showed that the nucleosome loci predominately (>75%) contained 1 or 2 labeled H2A copies (Figure 1E), again consistent with the notion that they were primarily loaded as spatially separated mononucleosomes. The other brighter foci contained more than 3 copies of labeled H4, likely representing multiple nucleosomes within the diffraction limit.
Nucleosome formation was also monitored by an increase in the tension along the tether when the positions of the OTs remained constant as more DNA was wrapped around histone octamers (Figure 1B, bottom). In the absence of Nap1, histone octamers can still bind DNA but remain mostly unwrapped, as the associated tension when the OT positions were held remained constant (Figure 1F). It is possible that non-specific binding of histone octamers to DNA occurs even in the presence of Nap1. In another experiment, the same protocol to form nucleosomes containing Cy3-labeled H2A was performed, which yielded similar results as those performed with LD655-labeled H4 (Figure 1G), indicating the assembly of intact octamers into nucleosomes.
Using the setup described in step 4 (Figure 2A), nucleosome unwrapping was visualized over time on a kymograph as the tether distance increased (Figure 2B). It was previously reported that histone octamers often remain bound to the DNA after being unwrapped30,31, and therefore, fluorescent trajectories often persist even at high tensions. Nucleosome unwrapping was also monitored on the associated FD curve (Figure 2C). Quantifiable features of the FD curve include the force at which nucleosome(s) unwrap (see "Transition F" in Figure 2C inset) as well as the length of DNA that was released from the histone octamer (see "ΔL" in Figure 2C inset), which exists in multiples of ~24 nm, consistent with independent unwrapping of the inner turns of DNA of individual nucleosomes27,30. These parameters can be used to estimate the number of nucleosomes formed on each tether or for additional downstream analysis of nucleosome mechanics. As such, the number of inner-turn ruptures obtained from mechanical pulling experiments was compared to the estimated number of labeled H4 copies at each nucleosome trajectory based on the monomer intensity, which showed that the numbers are largely consistent (examples shown in Figures 2D–G). However, it was noted that they do not perfectly match due to the ~70% labeled efficiency of the H4 protein in the sample and any residual Nap1-independent histone binding to DNA, as shown in Figure 1F. Finally, as expected, stretching Nap1-bound bare DNA tethers (without histones) do not generate force-induced transitions (Figure 2H), indicating that Nap1 alone does not significantly distort DNA.
Employing the setup described in step 5 (Figure 3A), the binding of linker histone H1 to chromatin was visualized as the appearance of green fluorescent foci along the chromatin tether (Figure 3B). Notably, H1-bound nucleosomes generated dual-color trajectories (see white arrows in Figure 3B) that appeared stationary over time. In addition, diffusing H1 trajectories across nucleosome-free DNA regions were observed (see green stars in Figure 3B).
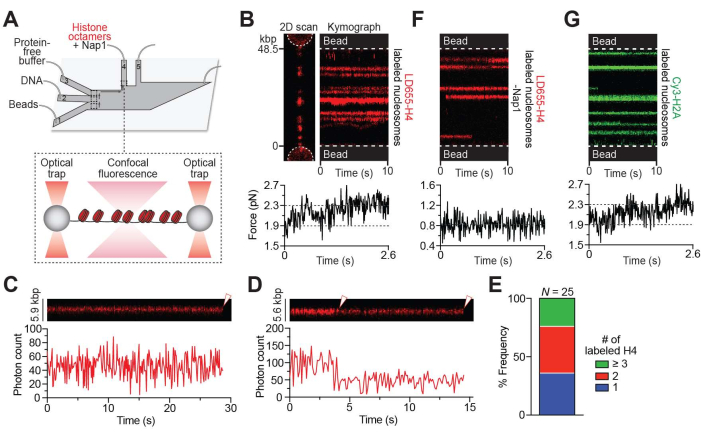
Figure 1: Nap1-mediated in situ nucleosome formation. (A) Schematic of the experimental setup described in the Nap1-mediated in situ nucleosome formation protocol (step 3). A pair of beads were optically trapped in ch. 1, and a single λ DNA molecule was tethered between the beads through biotin-streptavidin linkage in ch. 2. The tether was then moved to ch. 4, containing LD655-labeled histone octamers (red) and Nap1 for real-time formation of nucleosomes across the DNA. (B) Representative 2D scan (left) and kymograph (right) of an LD655-H4-labeled nucleosome-containing DNA tether. The associated force reading (bottom) shows an increase in force immediately as the tether is moved to ch. 4 when the OT positions are held constant, indicating nucleosome wrapping. Brighter trajectories signify multiple adjacent nucleosomes located within the diffraction limit of ~300 nm. (C) Example kymograph showing a 1-step photobleaching event (white arrow) of a nucleosome containing LD655-H4. (D) Example kymograph showing a 2-step photobleaching event (white arrows) of a nucleosome containing LD655-H4. (E) Relative frequency of the number of LD655-labeled H4 per trajectory estimated from their fluorescence intensity. (F) Representative kymograph of a tether bound with histone octamers in the absence of Nap1. The associated force reading (bottom) remains unchanged even as histone octamers bind the DNA. (G) Representative kymograph of a Cy3-H2A-labeled nucleosome-containing DNA tether. The associated force reading (bottom) shows an increase in force immediately as the tether is moved to ch. 4 (containing octamers and Nap1), as nucleosomes are formed when the OT positions are held constant. Please click here to view a larger version of this figure.
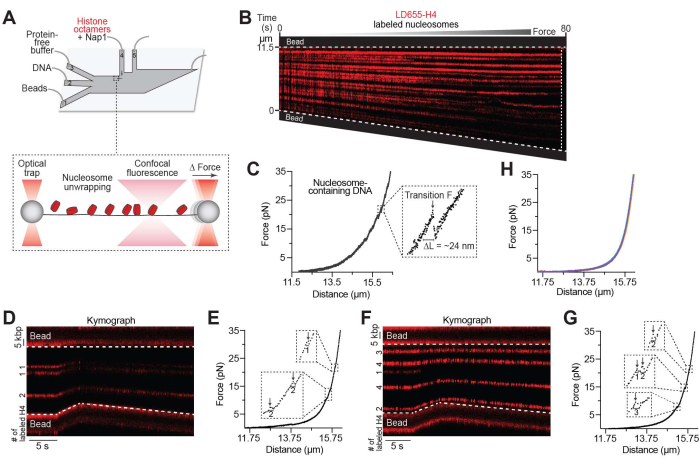
Figure 2: Nucleosome unwrapping assay. (A) Schematic of the experimental setup described in the unwrapping nucleosomes protocol (step 4). The DNA tether containing nucleosomes was moved to ch. 3 containing a protein-free image buffer where the inter-bead distance was increased and the nucleosomes were unwrapped. (B) Representative kymograph of a nucleosome-containing DNA tether pulled to high forces by gradually increasing the inter-bead distance. The decrease in fluorescence intensity toward the bottom bead was likely due to the DNA tilting slightly out of line scanning focus. (C) The associated FD curve showing force-induced transitions of inner turn unwrapping occurring between 8-37 pN. The inset shows a zoom-in view of an example transition for which the distance unwrapped (ΔL = ~24 nm for the inner turn of DNA of individual nucleosomes) and the transition force (Transition F) can be recorded and further analyzed. For this tether, 26 nucleosomes were unwrapped, and the tether ruptured at ~65 pN, as denoted by the white dotted line in the kymograph. (D,E) Example LD655-H4 nucleosome-containing tether pulled to high forces where the number of H4 per trajectory is estimated via fluorescence intensity (indicated in the kymograph, D) and the number of wrapped nucleosomes is estimated via the size of force-induced distance change (indicated in the associated force-distance curve, E). (F,G) Another example as in (D,E). (H) DNA tethers (n = 5) pulled to high forces after being incubated in a channel containing Nap1 only. Please click here to view a larger version of this figure.
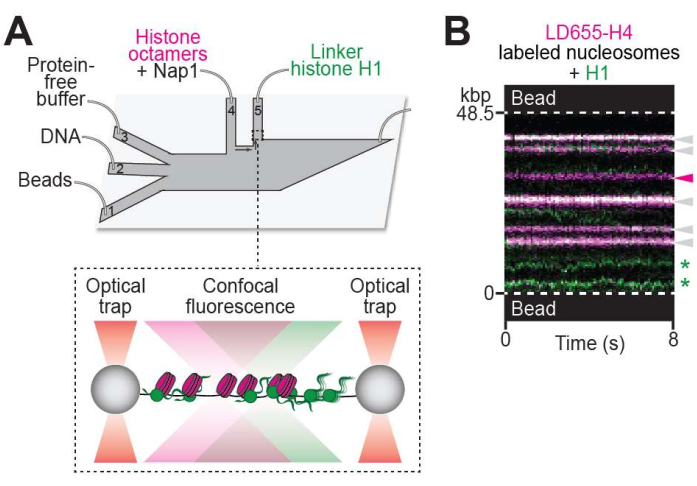
Figure 3: Visualizing protein-chromatin interaction. (A) Schematic of the experimental setup described in the visualizing protein binding to chromatin protocol (step 5). The DNA tether containing nucleosomes was moved to ch. 5, containing 10 nM of Cy3-labeled linker histone H1 (green). (B) Representative two-color kymograph of H1 binding to a nucleosome-containing DNA tether. H1-nucleosome colocalization events are denoted with white arrows. Unbound nucleosome trajectory is denoted with the magenta arrow. H1 molecules diffusing on bare DNA are denoted with green stars. Please click here to view a larger version of this figure.
