For cellular and lamella samples, the data collection strategy largely depends on the sample and the goal of the imaging study (Figure 1). The targeting approach depends on whether the molecular target is in situ or prepared from the purified macromolecular complex for high-resolution reconstruction specimens containing molecular targets. For purified complexes vitrified onto holey (carbon) grids, targeting can simply be based on imaging in the holes of the (carbon) support film. For in situ work, the targeting approach requires knowledge of the location of the molecular entity based on correlative data or known low-magnification cellular landmarks. Cellular landmarks would ideally be identifiable when taking overview images and, if sufficient to roughly localize regions of interest, can provide a quick way to confirm target identity with a search image. However, if the observable events are rare, then medium magnification search images may be needed to qualify the target is correct. Search maps are medium magnification montages of search images and can, thus, make target finding much easier, where they can be acquired at a magnification at which the feature of interest is visible. Search maps can then be screened to find and set up target batch positions. For specimens containing cellular features for ultrastructural reconstruction, the targeting approach is similar, though equally dependent on the visibility of the cellular event at various magnifications and its prevalence in the sample.
The data acquisition strategy must also be considered; in all cases, the study's goal largely determines how the data is collected. For reconstruction of the cellular ultrastructure, a low magnification (20-5 Å/px) and large field of view may be appropriate, but high magnifications are required to reconstruct molecular or high-resolution detail (5-1 Å/px). A dataset collected at 1.5 Å/px under ideal conditions will only physically be able to produce a reconstruction at the Nyquist frequency of 3.0 Å/px; however, in reality, many factors, including but not limited to specimen thickness, size, and heterogeneity, all affect the obtained reconstruction quality. The exact imaging parameters also balance the magnification based on the study's goal with the field of view to contain enough information. This article presents a subtomogram averaging case reaching 2.86 Å, but additional studies17,42,43,44,45 are presented in Table 1 to illustrate the collection parameters associated with studies targeting different outcomes17,42,45,46.
Once a targeting workflow and data acquisition regime for cryo-ET is established, data collection of many different sample types is possible. Representative tomograms of a variety of specimens are presented here: molecular samples such as apoferritin (Movie 1), thin cellular processes (Movie 2), and FIB milled lamella of the thick cellular specimen (Movie 3).
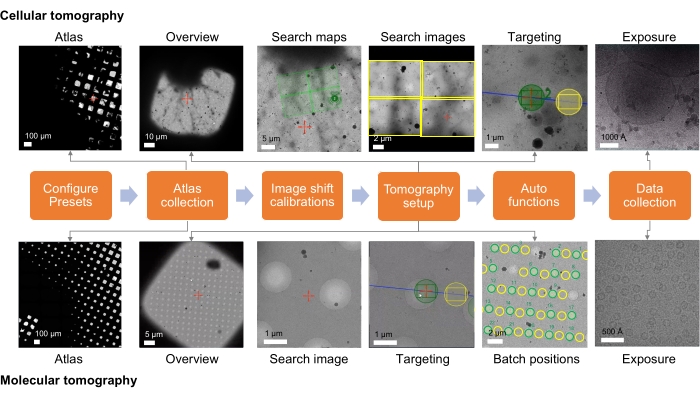
Figure 1: Overview of the tomography workflow setup. The cryo-ET imaging workflow described in the protocol is shown as a flow chart. The images expected to be acquired are shown for the cellular and molecular workflow. The presented naming follows the Tomo5 convention, although most tomography acquisition software shares common principles for collecting these images. Please click here to view a larger version of this figure.
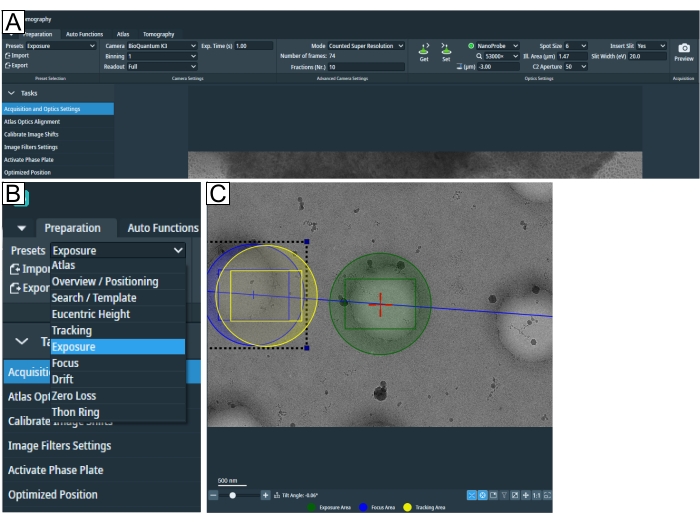
Figure 2: Preparation tab and search preset conditions. (A) Overview image of the whole tab. Imaging condition presets are set in this tab, and the "Calibrate Image Shift" and "Image Filter Settings" can be found in the "Tasks" drop-down. (B) Zoom-in of the "Presets" drop-down where each preset can be selected for individual imaging conditions setup. (C) Image depicting an adequate magnification to fit both the exposure and the "Focus and Tracking" area into the field of view. Please click here to view a larger version of this figure.
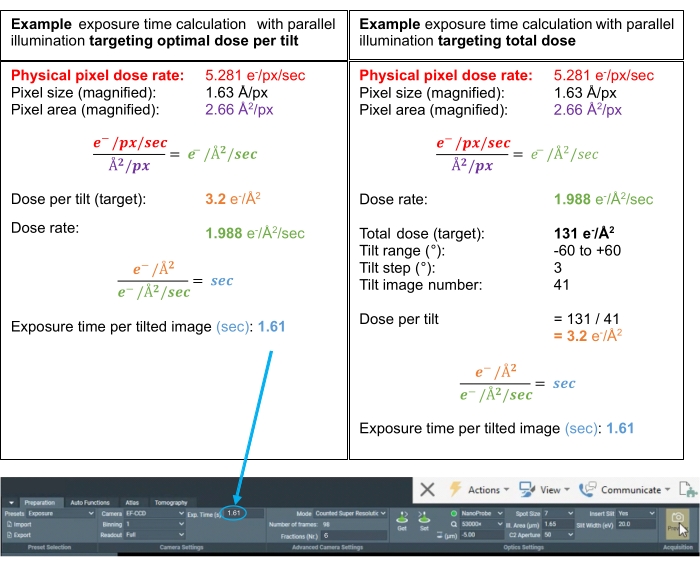
Figure 3: Dose calculations. Example dose calculations for possible tomogram acquisition schemes where the dose rate has been measured over vacuum. The two calculations determine the exposure time (s) for each tilt, whether targeting an optimal dose per tilt or an optimal total dose for the full tilt series. In subtomogram averaging, it is common practice to target an optimal dose per tilted image in the range of 3.0-3.5 e–/Å2. In both cases, the "Fractions (Nr.)" are set to 6 to achieve ~0.5 e–/Å2 of dose per movie frame of the tilt for sufficient signal to perform motion correction. Please click here to view a larger version of this figure.

Figure 4: Atlas tab. Overview image of the "Atlas" tab. The image has been cropped to avoid empty grid position spaces. The "Tasks" menu contains session setup preferences and grid selection spaces for individual selection of all grids inventoried and an option to acquire a single atlas after the cassette has been removed from the autoloader. A selected grid can be reset and then re-acquired. Please click here to view a larger version of this figure.

Figure 5: Calibrated image shifts. Image depicts an exposure and search image. The red cross in the search image is the shifted marker to correct the offset between exposure and search. One should redo image shift calibrations at the session start or after changing the imaging condition presets. Please click here to view a larger version of this figure.
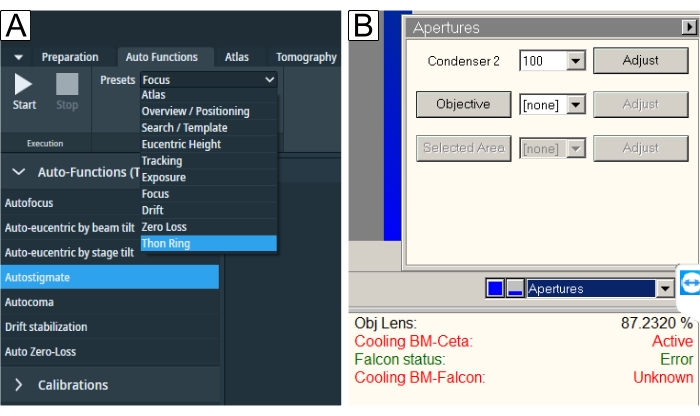
Figure 6: Auto functions and apertures. (A) The "Auto Functions" tab depicts the "Presets" drop-down and the "Task" selection. Underlined in blue is the "Thon Ring" preset required for "Autostigmate" (also underlined in blue) and "Autocoma". One should select respective presets for each task and then press the Start button. (B) Apertures are found in the TEM User Interface. Select desired "Objective aperture" after the auto functions have been performed and run "Autostigmate" with the aperture in. Please click here to view a larger version of this figure.
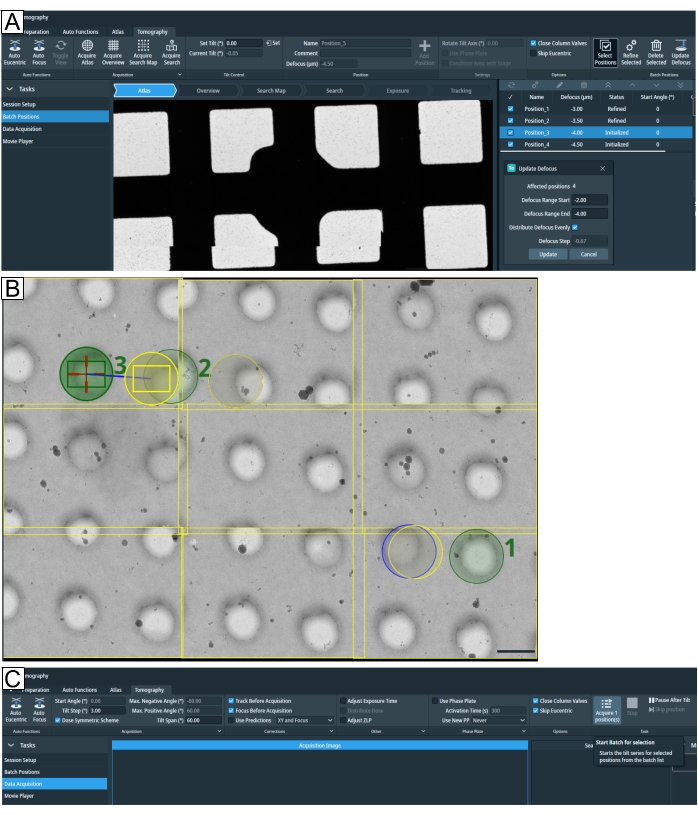
Figure 7: Tomography tab overview. Images show the Tomo 5.8 user interface. (A) Batch positions. The latest functions depicted in the image: the "Acquire Search Map" option; tomogram positions are shown in atlas view with the zoom-in option; highlighted at the top right is "Select Positions"; below all four positions have been selected to "Update Defocus" parameters. (B) Three positions are selected on the search map, labeled 1, 2, and 3. Position 1 will not pose a problem when "Refine All" is run. However, if in a high target density setting such as when lamellae positions are selected, as depicted for position 2 and position 3, then "Refine All" will run the "Tracking" and "Focus" routine on the "Exposure" region of position 2, exposing the target before the tomogram is acquired. The grid type is R1.2/1.3. Scale bar = 1.2 µm. (C) Data acquisition. Selected positions setup in "Batch Positions" can now be individually acquired. Please click here to view a larger version of this figure.
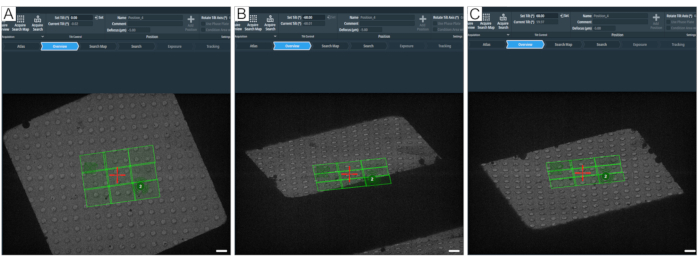
Figure 8: Defining the maximum tilt angle. Figure depicts a step-by-step approach to determining the maximum tilt range for tomogram acquisitions. (A) 0° overview and a search map in the center of the grid square. To test the tilt range, the angle can be set in the tomography software with "Set Tilt (°)" by typing the desired value and then pressing Set. (B) The stage has been tilted to −60°, showing that a corner of the search map would not be fully acquired at −60°. (C) The stage has been tilted to 60°. By counting the holes that have disappeared at ±60°, one can get an idea about the tilt range of a grid square. Scale bars = 2.5 µm. Please click here to view a larger version of this figure.
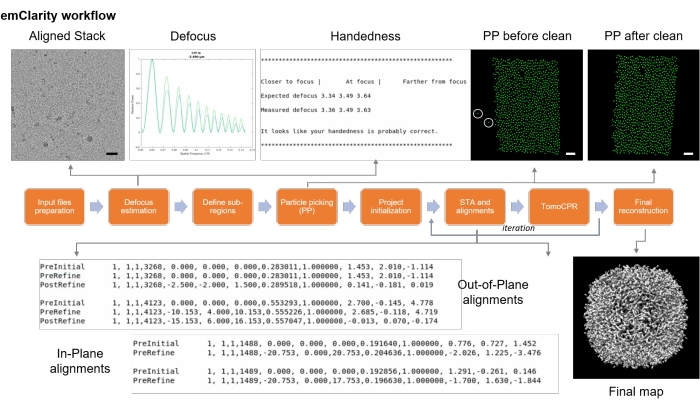
Figure 9: emClarity flowchart. The flowchart described the various steps for cryo sub-tomogram averaging. Scale bars = 50 nm. Please click here to view a larger version of this figure.

Figure 10: Template matching using emClarity. (A) A typical micrograph of apoferritin on a graphene-coated grid. Defocus: −3.430 µm. (B) A slice of the tomogram overlaid with model points after template searching. (C) A top and (D) a side projection views of model points, indicating a single flat layer of monodispersed apoferritin particles. Scale bars = 50 nm. Please click here to view a larger version of this figure.
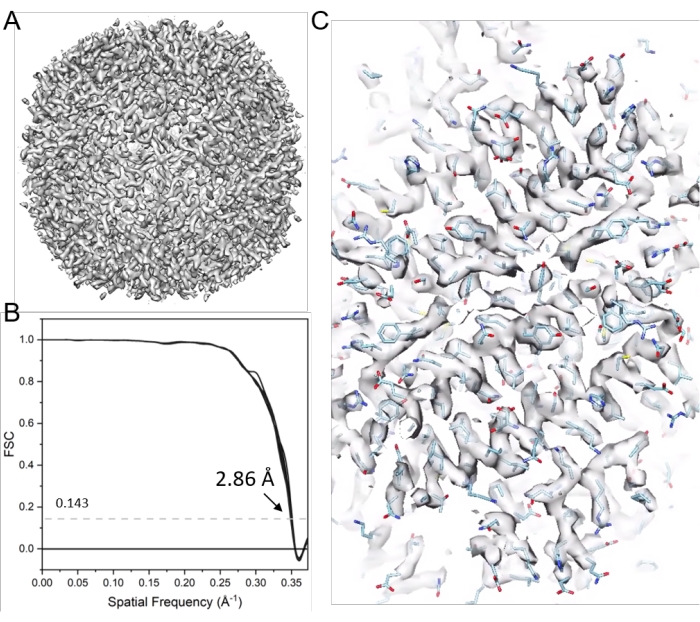
Figure 11: Cryo-ET STA of apoferritin. (A) The final map after 21 cycles of sub-tomogram alignment. (B) The final map's Fourier shell correlation (FSC) plot with a reported resolution of 2.86 Å, containing 38 cone FSCs. (C) Representative density maps (fitted with PDB model 6s6147). Please click here to view a larger version of this figure.
| Sample | Cryo-ET type | Å/px | Tilt range (+/-) | Tilt step (°) | Defocus range (µm) | Total dose (e-/Å2) | Resolution | Raw data | Reference |
| Apoferritin | Purified/Molecular (STA) | 1.34 | 60 | 3 | 1.5 – 3.5 | 102 | 2.86 | EMPIAR-10787 | 17 & this paper |
| HIV-1 Gag | Purified/Molecular (STA) | 1.35 | 60 | 3 | 1.5 – 3.96 | 120 | 3.1 | EMPIAR-10164 | 17 |
| Ribosome | Purified/Molecular (STA) | 2.1 | 60 | 3 | 2.2 – 4.3 | 120 | 7 | EMPIAR-10304 | 42 |
| SARS-CoV-2 spike | Lamella/Molecular (STA) | 2.13 | 54 | 3 | 2 – 7 | 120 | 16 | EMPIAR-10753 | 45 |
| Neuron axon structure | Cellular (ultrastructural) | 5.46 | 60 | 2 | 3.5 – 5 | 90 | N.D. | EMPIAR-10922 | 47 |
Table 1: Collection parameters for several cryo-ET studies. Studies targeting the reconstruction of molecular detail from purified or in situ proteins compared to a study aiming to resolve and segment the ultrastructural cellular features.
Movie 1: A tomogram of apoferritin samples on a normal EM grid and then imaged with a cryo-TEM equipped with an ultra-high-resolution camera with a compatible filter. Tilt series were acquired with a dose-symmetric scheme, with a tilt span of 54° and a total dose of 134 e–/A2 in the electron tomography software. Scale bar = 50 nm. Please click here to download this Movie.
Movie 2: A tomogram of a primary neuron grown on an EM grid and then directly imaged with a cryo-TEM equipped with an ultra-high-resolution camera with a compatible filter. The tilt series were acquired with a dose-symmetric scheme, with a tilt span of 60° and a total dose of 120 e–/A2 in the electron tomography software. Scale bar = 100 nm. Please click here to download this Movie.
Movie 3: A tomogram of a Cyanobacteria on an EM grid, subjected to FIB milling and then imaged with a cryo-TEM equipped with a high-speed camera and a compatible filter. The tilt series were acquired with a dose-symmetric scheme, with a tilt span of 50° and a total dose of 120 e–/A2 in the electron tomography software. Scale bar = 87.2 nm. Please click here to download this Movie.
Supplementary File 1: The parameter file template for estimating the defocus. Please click here to download this File.
Supplementary Table 1: Data collection and microscope setup details. Please click here to download this Table.
Supplementary Table 2: List of commands in the order of execution. Please click here to download this Table.
