Covalent coupling of peptide and lipid
Reaction completion and desired product formation was confirmed by TLC. A separate unreacted peptide control did not move up during TLC: it was retained at the start, and its spot was positive for the primary amino group, as observed after ninhydrin spray, upon heating. This ninhydrin-positive free peptide spot was no longer observed in the mixture following reaction completion, following TLC of the reaction mixture sample, after the removal of DIPEA, DMSO, and re-dissolution in chloroform. As for the crucial issue of NHS ester reagent quality, Figure 1 presents spectrophotometric track of hydrolysis kinetics, with the zero time point at the beginning of the reaction being when the NHS ester in an organic solvent was added to the cuvette. This confirms the functionality of NHS active ester of carboxy-PEG-DSPE (see Methods Section 1). At the zero time point, the extrapolated A260=0.33 represents the material that was already hydrolyzed prior to testing. At the completion of the hydrolysis reaction, in excess of 10-15 min, A260=1.54 (when absorbance does not increase considerably anymore). This confirms the presence of active ester. It also provides quantitative data, that over 78% of the material is not pre-hydrolyzed NHS, and can be thus successfully used for peptide coupling, with the proper adjustment of the reagent amount.
Preparation and transfer of lipid material from the aqueous media onto the bubble shell: fluorescence lipid
The microbubbles for this study were prepared to contain a trace amount (under 1%) of the fluorescent dye DiI, with characteristic red fluorescence, that was added as a solution in PG to the saline-PG solution of DSPC and PEG stearate. Resulting microbubbles clearly demonstrate shell fluorescence when green light excitation and red emission filters are used in the microscope (see Figure 2, left). Brightfield microscopy of microbubble gas phase (Figure 2, right) can be compared with microbubble shell fluorescence. For the quantitative assessment of lipid material transfer from the aqueous phase to the bubble shell, microbubbles were floated using centrifugation, and the fluorescence signal of the clear infranatant phase was compared with the fluorescence of the initial solution, prior to microbubble amalgamation. Almost an order of magnitude signal reduction was observed (Figure 3), i.e., over 85% of the lipid material has transferred to the microbubble shell by amalgamation.
Preparation and size distribution correction of microbubbles
Microbubbles generated by amalgamation demonstrated a typical size distribution, with high concentration (e.g., ~4.8 x 109 particles per mL for biotinylated bubbles). The size distribution was wide, with particles present within the measured range (between 1 and 30 µm); ~6.3% microbubbles exceed 5 µm in diameter (Figure 4, green curve). Intravascular administration of large microbubbles may lead to their nonspecific accumulation in the blood capillaries and should be avoided. A short (15-17 min) flotation of the inverted vial in normal gravity, with the subsequent collection of 0.3 mL close to the septum surface, allows removal of larger microbubbles completely, with minor loss in the total particle number concentration, down to ~4.6 x 109: following flotation, only 0.01% of the particles in the purified sample have diameters over 5 μm (Figure 4, red curve).
Adhesion of microbubbles to receptor-coated surface: static assay
This procedure has been first described in the previous century1, and is being used as a quick test that confirms functionality of targeted microbubbles. Microbubbles are allowed to contact the receptor-carrying dish surface. If the ligand-receptor interaction takes place, bubbles may be retained on the surface despite the vigorous wash. An example of such a quick test of the functional adhesion of c(RGDfK)-microbubbles onto the surface coated with recombinant αvβ3 is presented. Figure 5 is a representative brightfield microscopy image of adherent microbubbles on the receptor surface in a Petri dish, following a wash with PBS, to remove unbound bubbles. Bubbles in this type of microscopy present as dark circular patterns. In similar condition, if the surface is only coated with albumin (to block nonspecific adhesion), microbubbles will not adhere and will be easily washed away even by the gentle rinse.
Binding of microbubbles from the flowing medium: parallel plate flow chamber
This procedure has been initially proposed as a tool for study of cell adhesion in a controlled flow setting15 and adapted for study of microbubble targeting decades later11. Testing in a flow-through system, unlike a static assay, is much more realistic for a clinical imaging scenario, where circulating bubbles in a flow of blood briefly touch the vessel wall and may adhere to it if the target receptor is present. Two examples of such studies are presented. The first example is a more traditional approach, where the adhesion of peptide-decorated microbubbles to the receptor-coated surface is monitored by video microscopy. Microscopy allows distinguishing adherent microbubbles from the flowing ones. It also allows to quantify those adherent microbubbles in the microscope imaging frame: many more c(RGDfK) microbubbles (left column) adhere to the surface, when compared with control, where scrambled c(RADfK) peptide is used, or if the surface is only coated with BSA (Figure 6).
The second example is contrast ultrasound imaging of the Petri dish coated with streptavidin (Figure 7, right side) to which biotinylated microbubbles adsorb successfully from the flowing medium, and can be detected by contrast ultrasound imaging following a flush with PBS. The control dish surface does not retain any adherent microbubbles from the flow, so essentially all ultrasound contrast signal is removed with PBS flow. Ultrasound contrast signal quantification shows strong statistical significance of the difference observed; the ratio of target and control signals exceeded an order of magnitude.
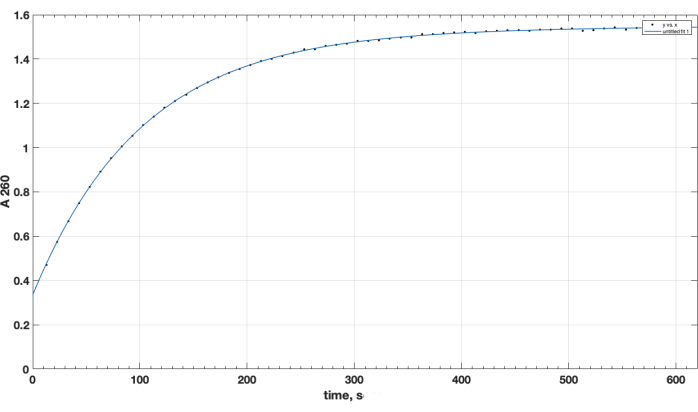
Figure 1. Kinetics of hydrolysis of NHS-PEG-DSPE active ester, observed by NHS release in alkaline medium by spectrophotometric testing at 260 nm wavelength. Zero time point is the time of addition of NHS-PEG-DSPE in organic solvent to the 0.1 M borate buffer, pH 9.2. Please click here to view a larger version of this figure.

Figure 2. Microscopy of gas-filled microbubbles following amalgamation. Left, fluorescence microscopy (green excitation, red emission, DiI lipid shell dye). Right, brightfield microscopy (gas phase observation), same magnification. Frame width, 85 um (10 μm stage micrometer embedded on bottom right of each image). Please click here to view a larger version of this figure.
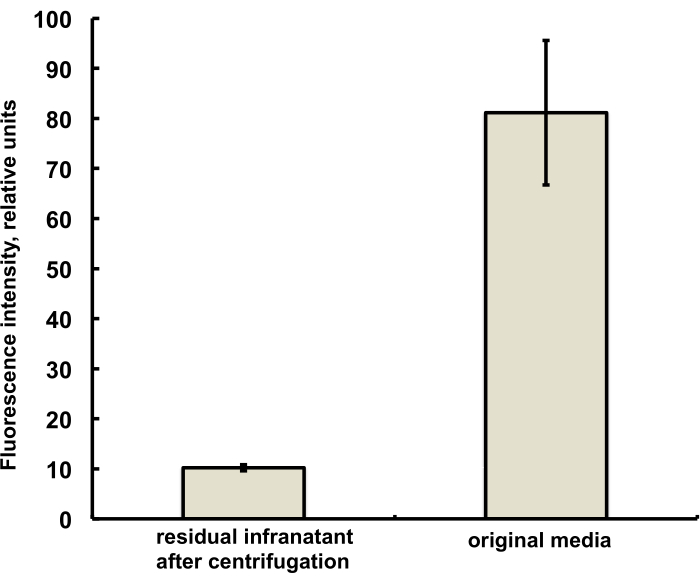
Figure 3. Fluorescence spectroscopy of DiI lipid dye sample from the microbubble preparation medium before amalgamation (right) and following amalgamation and removal of microbubbles by centrifugal flotation (left). Fluorescence excitation – 555 nm, emission – 620 nm. Data presented as Mean ± Standard Deviation. Please click here to view a larger version of this figure.
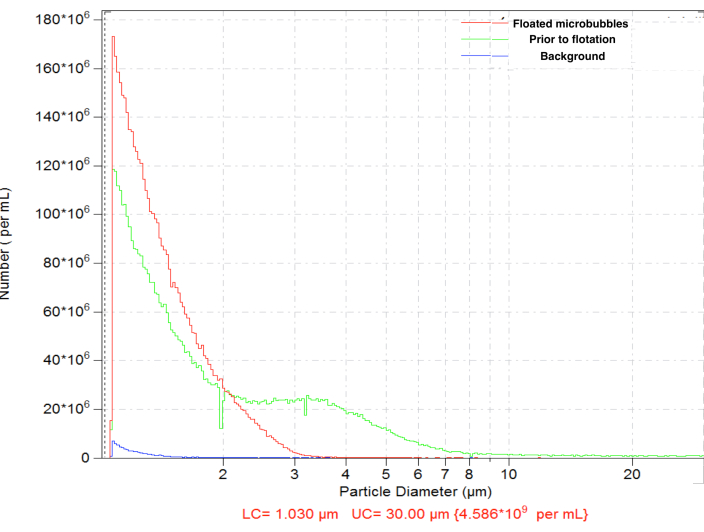
Figure 4. Particle size distribution of the number concentration of microbubbles following amalgamation preparation (green), with subsequent normal gravity flotation for the removal of large microbubbles (red) and diluent-only background count (blue). Electrozone sensing particle counting in normal saline, 50 µm orifice. Please click here to view a larger version of this figure.

Figure 5. Brightfield microscopy of c(RGDfK)-microbubbles on a dish coated with αvβ3. Image frame width is 106 μm; bar is 10 μm. Please click here to view a larger version of this figure.
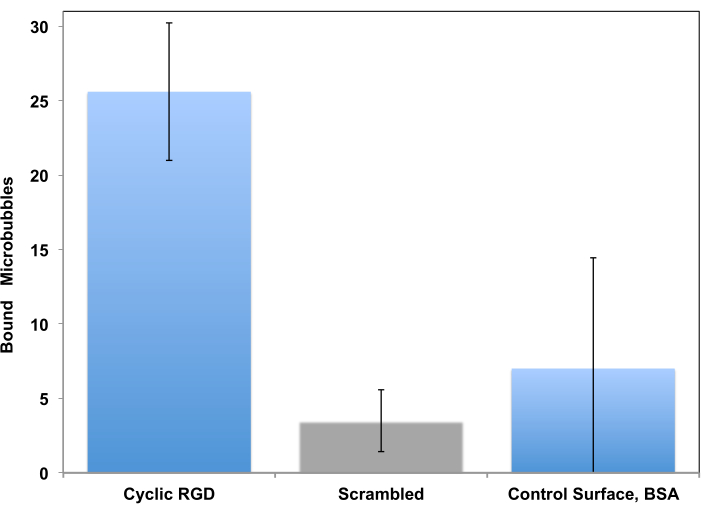
Figure 6. In vitro parallel plate flow chamber targeting of peptide-decorated microbubbles to the surface coated with recombinant αvβ3. cRGDfK-decorated microbubbles efficiently adhered to the dish (left), attachment of control non-targeted cRADfK (scrambled, center) microbubbles was minimal (p<0.00005), as was microbubble retention at the albumin-only control surface (right, p<0.0025). Chamber flow wall shear stress at 1 dyn/cm2. Microbubble adhesion monitored by video microscopy; the number of particles in the field of view is presented. Accumulation time is 4 min. Data presented as Mean ± Standard Deviation. Reprinted with permission from5. Copyright, 2018, American Chemical Society. Please click here to view a larger version of this figure.
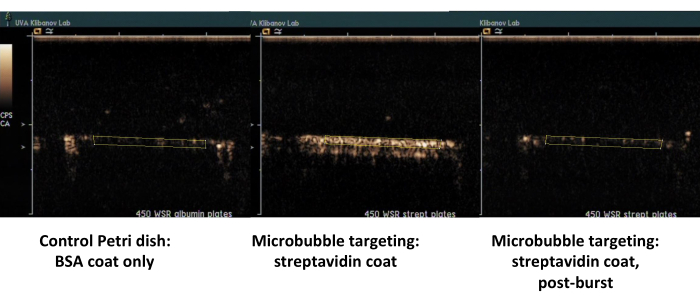
Figure 7. Contrast ultrasound imaging of a parallel plate flow chamber following targeted adhesion and buffer flush of biotinylated microbubbles on the dish coated with streptavidin (middle, adherent targeted microbubbles, right, same dish, following high-MI ultrasound burst), and control dish coated only with albumin (left). Two minutes of perfusion of microbubble dispersion (PBS/BSA, 106 particles/mL) at 450 s-1 shear rate, followed by buffer flush. Quantification of ultrasound signal is performed from the regions of interest in the video frames after background subtraction. Please click here to view a larger version of this figure.
