Combined O3 and LPS exposure leads to systemic inflammation and bone marrow mobilization at 72 h: Cell counts in different compartments revealed significant changes in peripheral blood and the femur bone marrow total cell counts upon combined O3 and LPS exposures. Although combined O3 and LPS exposures did not induce any changes in the total BAL (Figure 1A) or LVP (Figure 1B) cell counts, polymorphonuclear cells presented as the predominant cell type at 24 (Figure 1F, Supplemental Figure 1), 36 and 72 h after exposure. Please note the absence of Siglec-f and CX3CR1 staining in polymorphonuclear cells at 24 h BAL cytospin image (Figure 1F). Triple DAPI/CD11b/Gr1 staining of BAL cytospins showed presence of large CD11b- and Gr1-positive mononuclear cells which are macrophages at 0 h, which changes to smaller polymorphonuclear cells showing punctate Gr1staining but devoid of CD11b staining at 4 h (as shown in the inset in Supplemental 1F). The majority of BAL cells are polymorphonuclear and positive for both CD11b and Gr1, at 24 h post exposure (Supplemental Figure 1).
The mice displayed systemic leukocytosis at 24 h (3.3-fold vs 0 h, p<0.05, Figure 1C) followed by leukopenia at 72 h which was marked by13-fold lower counts in peripheral blood compared to 24 h (p<0.01) and a 8.6-fold lower compared to 4 h (p<0.05) after combined O3 and LPS exposure (Figure 1C). The sternal bone marrow cells were also unchanged when compared to 0 h or after combined O3 and LPS exposure (Figure 1D). The femur bone marrow counts showed a late 28.8-fold spike at 72 h when compared to 0 h (p<0.01, Figure 1E) as well as other time points (p<0.01, Figure 1E). Visualization of the LVP, and sternal as well as femur bone marrow cells also showed changes in the cell types to predominantly polymorphonuclear cells (Supplemental Figure 1). The sternal bone marrow cells showed signs of damage as evidenced by extracellular nuclear material. The femur bone marrow showed higher numbers of CD11b and Gr1 positive cells (Supplemental Figure 1), which coincide with the cell counts.
BAL total protein content was highest at 36 h after combined O3 and LPS exposure: Quantification of the total proteins in the LVP and BAL compartments was carried out to correlate ozone induced lung edema i.e., plasma protein exudation due to resulting vascular and epithelial barrier impairment due to the combined exposure. BAL total protein was 4.5-fold higher at 36 h (p<0.05) when compared to 4 h (Figure 2A). However, LVP protein was unchanged after exposure (Figure 2B).
Combined O3 and LPS exposure induces strong chemokine gradients in the lung vascular compartment and not BAL: Chemokine multiplex assays were conducted on both BAL and LVP supernatants. Please note that the relative differences between these two compartments represents preferential retention in a particular compartment. The eosinophil chemoattractant eotaxin-2 was 4.3-fold higher at 4 h in the LVP compartment compared to 0 h (p<0.05, Figure 3A). In the BAL compartment, eotaxin-2 was 3.2-fold lower at 4 h when compared to 0 h (p<0.01, Figure 3B). The BAL eotaxin-2 levels kept on decreasing consistently until 72 h with 12.6-fold reduction compared to 0 h (p<0.01, Figure 3B). The lymphocyte chemoattractant IL-2 was 10.1-fold higher at 4 h in the LVP compartment compared to 0 h (p<0.05, Figure 3C). In the BAL compartment, IL-2 was 5.0-fold lower at 36 h when compared to 0 h (p<0.05, Figure 3D). The BAL eotaxin-2 levels kept on decreasing consistently 5.9-fold reduction at 72 h compared to 0 h (p<0.05, Figure 3D).
Interestingly, many chemokines were altered at 4 h, in the LVP and not the BAL. Most of these chemokines showed modest, yet significant, increase in LVP levels at 4 h when compared to later time-points. Although eotaxin-1 levels were not high after the exposures when compared to 0 h, but the levels were significantly high at 4 h when compared to 24 (2.7-fold, p<0.05) and 72 h (5.0-fold, p<0.01, Figure 4A) after combined exposure. The levels of LVP TNFα were 3.4-fold higher at 4 h when compared to 36 h (p<0.05, Figure 4B). Similarly, LVP IFNγ was 2.9-fold higher at 4 h when compared to 72 h (p<0.05, Figure 4C). CX3CL1 levels were also high at 4 h when compared to 24 (1.7-fold, p<0.05), 36 (1.6-fold, p<0.05) and 72 h (2.2-fold, p<0.01) after combined exposures (Figure 4D). CCL27 levels showed higher levels at 4 h as well when compared to 24 (2.7-fold, p<0.05), 36 (3.9-fold, p<0.01) and 72 h (2.9-fold, p<0.05) after combined exposures (Figure 4E).
Some chemokines had a strong presence in the LVP at 4 h after combined exposures, and not changed in the BAL before and after exposure, , indicating a strong chemokine response from the pulmonary capillaries. At 4 h, IL-16 LVP levels were 3.4-fold compared to 0 h (p<0.01), 3.2-fold compared to 24 h (p<0.01), 4.5-fold compared to 36 h (p<0.01) and 4.6-fold compared to 72 h (p<0.01) after combined exposures (Figure 4F). At 4 h, the neutrophil chemokine, CXCL5 LVP levels were 2.0-fold compared to 0 h (p<0.01), 4.7-fold compared to 24 h (p<0.01), 2.2-fold compared to 36 h (p<0.01) and 2.7-fold compared to 72 h (p<0.01) after combined exposures (Figure 4G). At 4 h, CXCL10 LVP levels were 2.3-fold compared to 0 h (p<0.01), 2.1-fold compared to 24 h (p<0.05), 2.5-fold compared to 36 h (p<0.01) and 2.7-fold compared to 72 h (p<0.01) after combined exposures (Figure 4H). At 36 h, the neutrophil chemokine, SDF1α LVP levels were 4.2-fold compared to 0 h (p<0.01), 2.9-fold compared to 24 h (p<0.05), 6.8-fold compared to 72 h (p<0.01) after combined exposures (Figure 4I). At 4 h, MIP3β LVP levels were 2.3-fold compared to 0 h (p<0.05), and 2.3-fold compared to 72 h (p<0.05) after combined exposures (Figure 4J).
Combined O3 and LPS exposure induces necrosis, disrupts cellular cytoskeletal, plasma membrane and mitochondrial integrity: As the compartmental leukocyte counts and protein contents were not correlating with the LVP chemokine gradients, it became more important to visualise the cells obtained from these compartments. Ex vivo actin/tubulin staining of baseline (0 h) BAL cells showed presence of cortical actin, stress fibers as well as lamellipodia (shown in green, Figure 5 left upper panel) and microtubule network (shown in red, Figure 5 left upper panel) that coincided with reduced mitotracker staining indicating presence of active mitochondria (shown in red, Figure 5 left lower panel). More than 95% of the BAL cells were mononuclear at baseline. At 4 h, BAL cells showed extracellular nuclear material, punctate cytoplasmic actin staining devoid of lamellipodia and reduced cortical actin staining (shown in green, Figure 5 right upper panel),fanned microtubule network (shown in red, Figure 5 right upper panel) that did not correspond to the reduced mitotracker staining (shown in red, Figure 5 left right panel). DAPI normalized actin fluorescent intensity analysis showed a 3.7-fold decrease in the ratio indicating reduction in actin staining at 4 h after exposure (p<0.01, Figure 6A). Similarly, DAPI normalized tubulin fluorescent intensity also reduced by 1.5-fold after exposure (p<0.01, Figure 6B), indicating a reduction in tubulin staining. The loss of lamellipodia was evident with an increase in the circularity of BAL cellular cytoskeleton immediately i.e., at 2 h (p<0.01, Figure 6C) and 4 h (p<0.05, Figure 6C) after exposure when compared to 0 h and a decrease in the cytoskeletal circularity at 24 (p<0.05) and 36 h (p<0.01) after exposure, when compared to 0 h (Figure 6C).
Up to 24 h, the BAL cells from the combined exposure were double positive for calcein (in green, Figure 7) indicating presence of active esterase activity, and ethidium homodimer (in red, Figure 7), indicating that although some cells were dead (red), majority were partially compromised cells. At 36 h, the BAL cells were largely viable (green), indicating replacement of the compromised cells by leukocytes recruited from other systemic compartments (Figure 7).
Specific ATPα and Ly6G staining of the BAL cells revealed discrete intracellular localization (Movies 1 and 2) of both the proteins in alveolar macrophages as well as neutrophils after exposure (Figure 7). The combined exposure led to reduction in the DAPI normalized fluorescent intensity ratio of ATPα (Figure 7, 8A) and an increase in intracellular Ly6G protein content (Figure 7, Figure 8B, Movies 1 and 2). Reduction in ATPα staining, at 24 h after exposure, correlated with lower tubulin and to some extent reduced mitotracker staining. We also observed anuclear ATPα positive cell bodies after exposure, which could be platelets. However, we did not confirm our findings. At 2 h, we observed smaller mononuclear BAL cells (p<0.01 Figure 8C, p<0.01 Figure 8D) when compared to 0 h but at 4 h, the mononuclear cells were larger (p<0.01 Figure 8C, p<0.01 Figure 8D) when compared to 0 h. At 24 (p<0.01 Figure 8C, p<0.01 Figure 8D) and 36 h (p<0.01 Figure 8C, p<0.01 Figure 8D), the BAL cells became progressively smaller owing to a shift towards polymorphonuclear cells, when compared to 0 h.
Immunophenotyping of BAL cells revealed transient expressions of NK1.1, ATP β and Ki-67 and sustained expressions of Gr1, CX3CR1 and angiostatin positive BAL cells after combined O3 and LPS exposures: Immunofluorescent staining of the first set of BAL cytospins were normalized to DAPI staining. There was an increase in the cellular NK1.1 positive cells at 24 h (p<0.05 vs 0 h, Figure 9, 10A). At 36 h, the BAL cells had lower cellular NK1.1 fluorescent intensity (p<0.01 vs 0 and 24 h, Figure 9, 10A). The cellular Gr1 fluorescent intensity was higher at 24 h (p<0.01 vs 0 h, Figure 9, 10B) as well as 36 h (p<0.01 vs 0 h, Figure 9, 10B). Similarly, the cellular CX3CR1 fluorescent intensity was higher at 24 h (p<0.01 vs 0 h, Figure 9, 10C) as well as 36 h (p<0.01 vs 0 h, Figure 9, 10C).
When normalized to cellular CD61, which is also ubiquitous in expression, revealed an increase in the cellular ATPβ fluorescent intensity at 24 h (p<0.01 vs 0 h, Figure 9, 10D) and a decrease in the cellular ATPβ fluorescent intensity at 36 h (p<0.01 vs 0 and 24 h, Figure 9, 10D). Notably, ATPβ showed predominantly nuclear localization at 24 h compared to peripheral localization at 36 h (Figure 9). The cellular fluorescent intensity of BAL Ki-67, an indicator of cellular proliferation, was higher at 24 h (p<0.01 vs 36 h, Figure 9, 10E) compared to 0 and 36 h. The levels were below baseline levels at 36 h (p<0.01, Figure 9, 10E), most likely due to higher polymorpho-nuclear CD61 vs Ki-67 expression at 36 h (Figure 9). Lastly, the cellular fluorescent intensity of BAL angiostatin was consistently high at both 24 and 36 h (p<0.01 vs 0 h, Figure 9, 10F), indicating a specific increase in this metalloproteinase cleaved plasminogen fragment, during the lung inflammatory response.
Combined exposures produce wide-spread lung damage: Lung histology revealed that the combined exposures produce prolonged damage to lungs as seen in H&E stained cryosections (Figure 11). Although bronchiolar and alveolar septal damage was expected, it was surprizing to observe endothelial damage of larger vessels, at 36 h after exposure (Figure 11). There were adherent intravascular leukocytes, patches of leukocyte aggregates (including neutrophils) in the damaged alveolar septal and peri-bronchiolar regions (Figure 11).
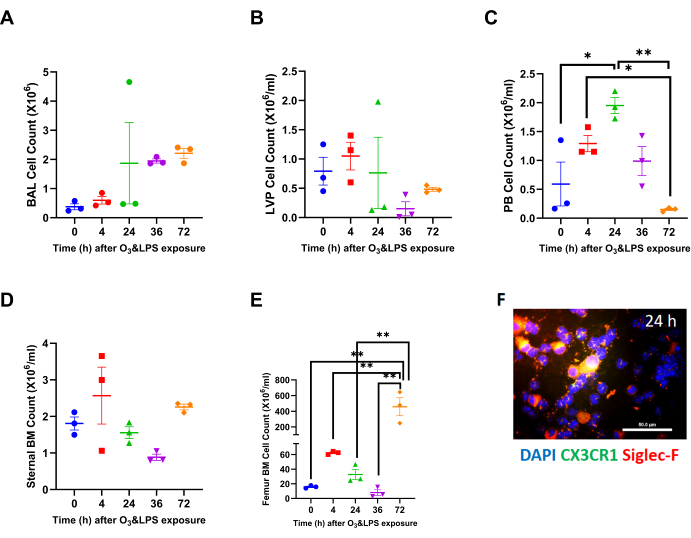
Figure 1: Compartmental leukocyte counts: A) Broncho-alveolar lavage (BAL) total leukocyte counts at 0 (i.e., baseline), 4, 24, 36 and 72 h after combined 0.05 ppb ozone (O3) and 50 µg intranasal LPS exposure to mice. B) Lung vascular perfusate (LVP) total leukocyte concentration at 0 (i.e., baseline), 4, 24, 36 and 72 h after combined 0.05 ppb ozone (O3) and 50 µg intranasal LPS exposure to mice. C) Peripheral blood (PB) total leukocyte concentration at 0 (i.e., baseline), 4, 24, 36 and 72 h after combined 0.05 ppb ozone (O3) and 50 µg intranasal LPS exposure to mice. D) Sternal bone marrow (BM) total leukocyte concentration at 0 (i.e., baseline), 4, 24, 36 and 72 h after combined 0.05 ppb ozone (O3) and 50 µg intranasal LPS exposure to mice. E) Femur bone marrow (BM) total leukocyte concentration at 0 (i.e., baseline), 4, 24, 36 and 72 h after combined 0.05 ppb ozone (O3) and 50 µg intranasal LPS exposure to mice. For the graphs A-E, 0 h is represented in blue, 4 h in red, 24 h in green, 36 h in purple and 72 h in orange data points; * p<0.05 and ** p<0.01. F) Representative BAL cytospin slide from 24 h exposure, showing mononuclear and polymorphonuclear cells when stained for nuclei with DAPI (in blue), CX3CR1 (in green) and Siglec-F (in red). Note that the merge of CX3CR1 and Siglec-F shows up as orange-yellow. Majority of mononuclear cells are positive for Siglec-F, which is a characteristic of alveolar macrophages. Scale bar = 50 µm. Please click here to view a larger version of this figure.
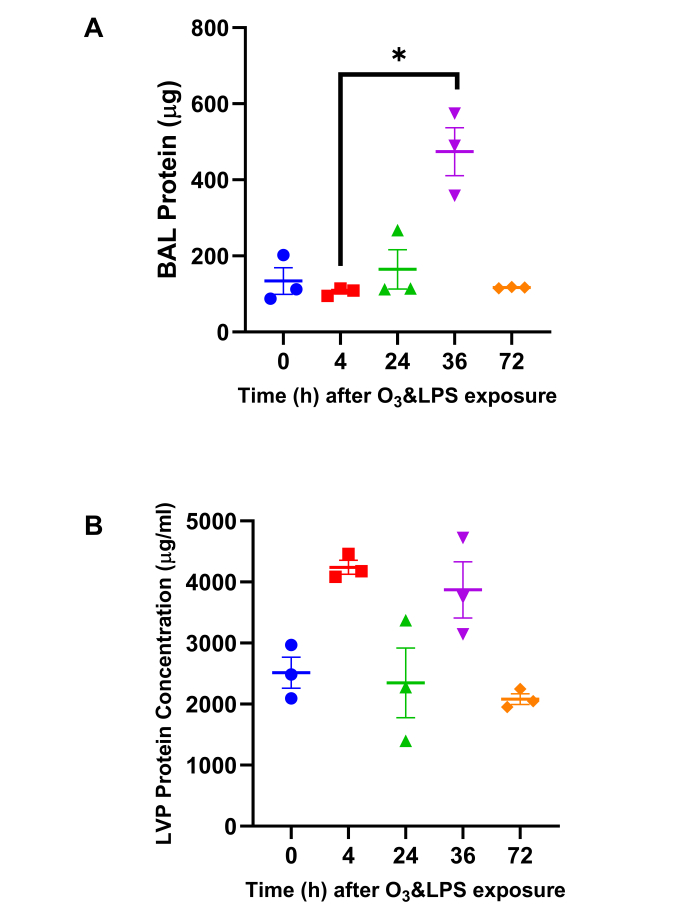
Figure 2: Total Protein quantification: A) Bronchoalveolar lavage (BAL) total protein content and B) Lung vascular perfusate (LVP) total protein concentration was estimated byPierce 660 nm protein assay (Thermoscientific, IL, US). For the graphs A-B, 0 h is represented in blue, 4 h in red, 24 h in green, 36 h in purple and 72 h in orange data points; * p<0.05. Please click here to view a larger version of this figure.
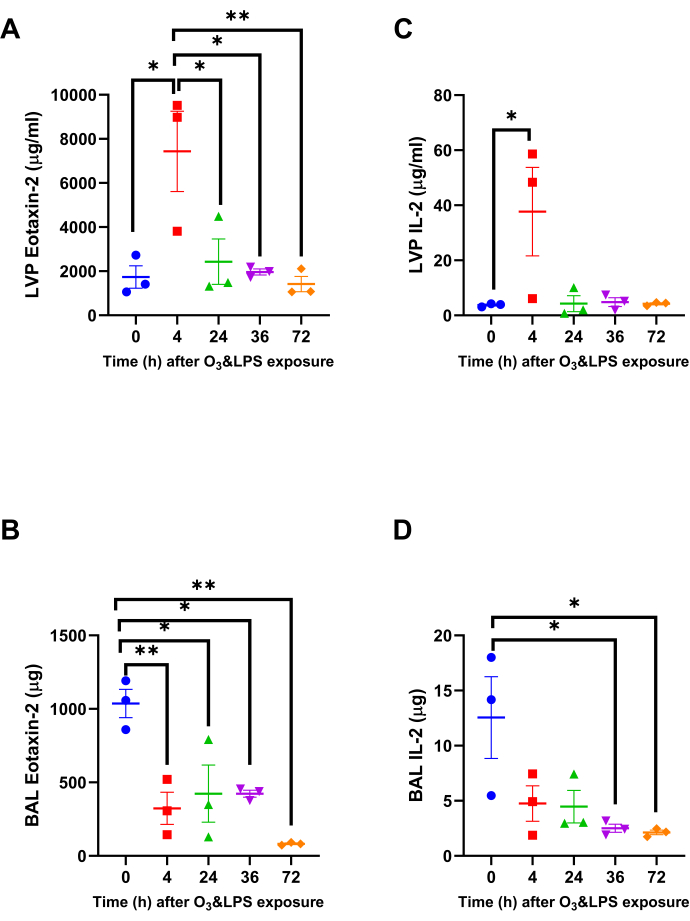
Figure 3: Lung Eotaxin-2 and IL-2 chemokine gradients: Out of the 33 chemokines, two chemokines that were significantly altered in the lung vascular perfusate (LVP) and bronchoalveolar lavage (BAL) and fluid after combined 0.05 ppb ozone (O3) and 50 µg intranasal LPS exposure to mice A) LVP eotaxin-2, B) BAL eotaxin-2, C) LVP IL-2 and D) BAL IL-2. Data were analyzed by one-way anova and the p-value for false discovery rate of multiple variables was adjusted as per Benjamini and Hoshberg correction. For the graphs A-D, 0 h is represented in blue, 4 h in red, 24 h in green, 36 h in purple and 72 h in orange data points. Pair-wise comparisons were analyzed after Bonferroni's correction. * represents p<0.05, ** represents p<0.01. Please click here to view a larger version of this figure.
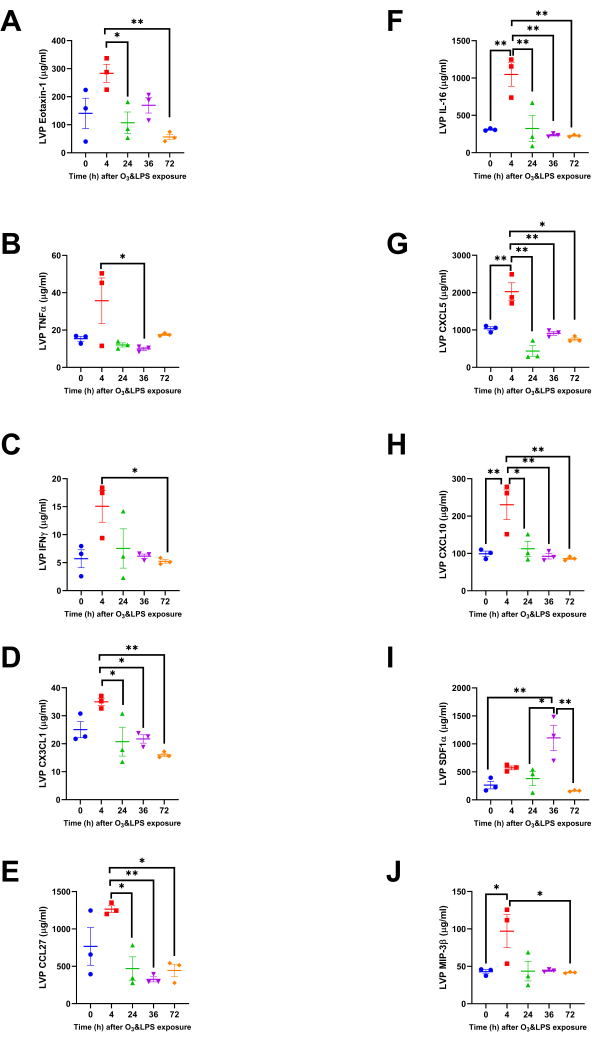
Figure 4: Lung vascular perfusate chemokine concentrations: Concentrations of ten more chemokines were altered, but only in the lung vascular perfusate (LVP) compartment. A) Eotaxin-1, B) TNFα, C) IFNγ, D) CX3CL1, E) CCL27, F) IL-16, G) CXCL5, H) CXCL10, I) SDF1α and J) MIP3β. For the graphs A-J, 0 h is represented in blue, 4 h in red, 24 h in green, 36 h in purple and 72 h in orange data points. Pair-wise comparisons were analyzed after Bonferroni's correction. * represents p<0.05, ** represents p<0.01. Please click here to view a larger version of this figure.

Figure 5: Bronchoalveolar lavage (BAL) cytospin actin/tubulin and reduced mitotracker staining: Representative images ofactin/tubulin stained BAL cytospins from A) 0 h and B) 4 h after combined 0.05 ppb ozone (O3) and 50 µg intranasal LPS exposure to mice. Note that DAPI is shown in blue, actin in green and tubulin in red. Representative images ofreduced mitotracker stained BAL cytospins from A) 0 h and B) 4 h after combined 0.05 ppb ozone (O3) and 50 µg intranasal LPS exposure to mice. Note that DAPI is shown in blue, and reduced mitotracker in red. Scale bar = 50 µm. Please click here to view a larger version of this figure.
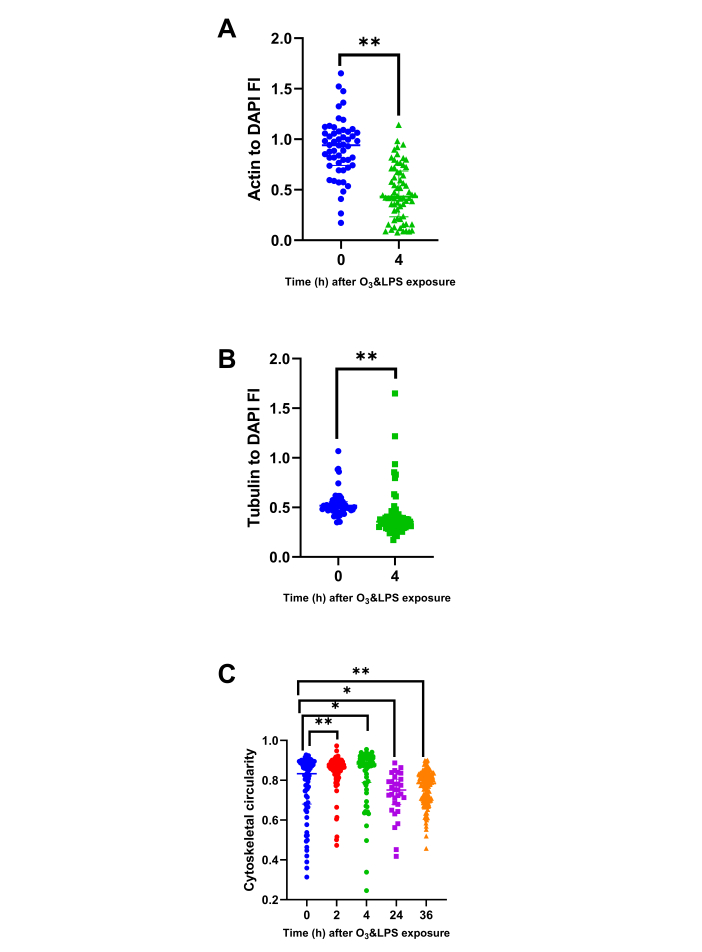
Figure 6: Bronchoalveolar lavage (BAL) cytoskeletal protein and circularity analysis: BAL cytospins stained for actin and tubulin were analyzed for DAPI normalized parameters i.e., A) Actin to DAPI fluorescent intensity (FI) ratio at 0 and 4 h, B) Tubulin to DAPI fluorescent intensity (FI) ratio at 0 and 4 h and C) BAL cell cellularity at 0 (n=75 cells), 2 (n=105 cells), 4 (n=66 cells), 24 (n=31 cells), 36 (n=154 cells) h. As a few pilot experiments were also performed immediately after O3 and LPS exposures i.e., at 2 h time-point, we also included some very early BAL cellularity analysis from the 2 h time-point to highlight the immediate effects of O3. For the graphs A-C, 0 h is represented in blue, 2 h in red, 4 h in green, 24 h in purple and 36 h in orange data points. * represents p<0.05, ** represents p<0.01. Please click here to view a larger version of this figure.

Figure 7: Bronchoalveolar lavage (BAL) intracellular and plasma membrane status: BAL cells were stained for vital stains, calcein (in green) and ethidium homodimers/EthHD (in red) as shown in representative upper image panels at A) 0, B) 24 and C) 36 h after O3 and LPS exposures. Scale bar = 200 µm. BAL cytospins were immunostained for DAPI (in blue), ATPα (in green) and Ly6G (in red). Representative images are shown in the lower image panels at A) 0 and B) 24 h after O3 and LPS exposures. Scale bar = 50 µm. Please click here to view a larger version of this figure.
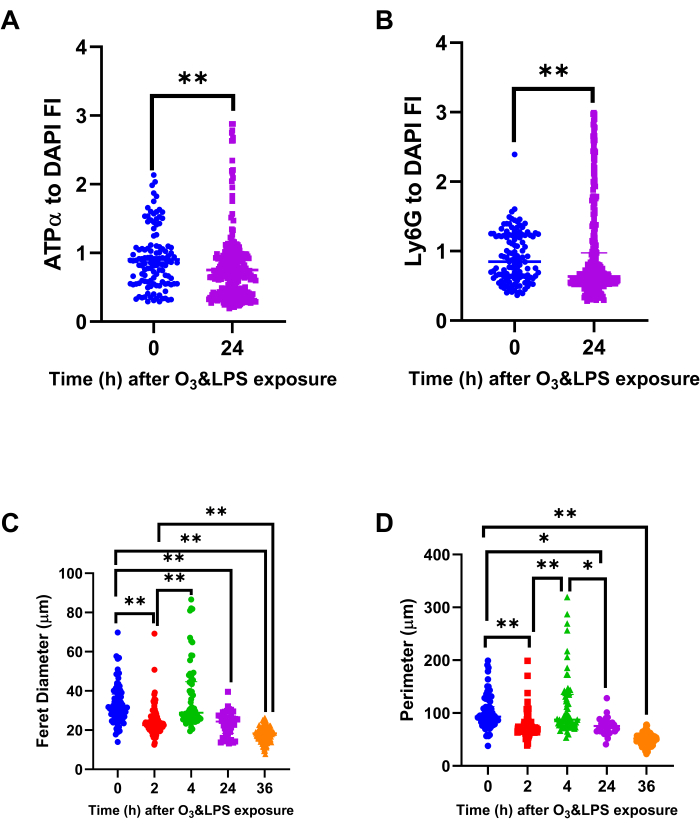
Figure 8: Bronchoalveolar lavage (BAL) mitochondrial protein ATPα, lysosomal protein Ly6G and cell size analysis: Immunostained BAL cells were normalized for DAPI to compute A) ATPα to DAPI fluorescent intensity (FI) ratio and B) Ly6G to DAPI FI ratio, at 0 and 24 h after O3 and LPS exposures. Immunostained BAL cells were also analyzed for their A) longest i.e., ferret diameter and B) perimeter, at 0, 2, 4, 24 and 36 h after O3 and LPS exposures. For the graphs A-D, 0 h (n=119 cells) is represented in blue, 2 h (n=105 cells) in red, 4 h (n=66 cells) in green, 24 h (n=309 cells) in purple and 36 h (n=154 cells) in orange data points. * represents p<0.05, ** represents p<0.01. Please click here to view a larger version of this figure.
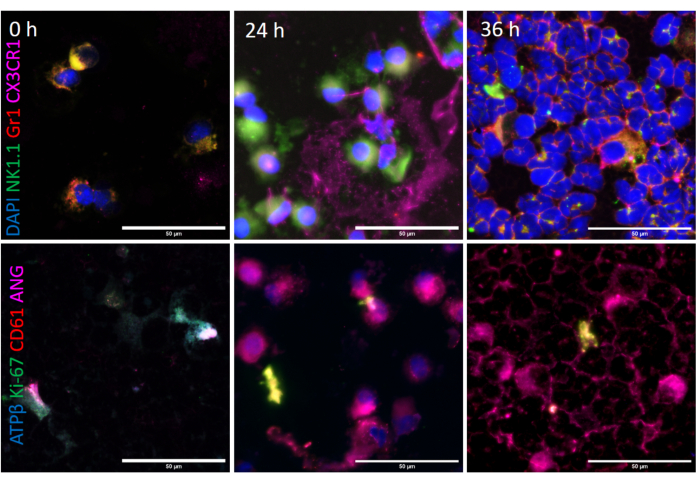
Figure 9: Bronchoalveolar lavage (BAL) intracellular phenotyping: BAL cells were immunostained for DAPI (in blue), NK1.1 (in green), Gr1 (in red) and CX3CR1 (in magenta) as shown in representative upper image panels at A) 0, B) 24 and C) 36 h after O3 and LPS exposures. Scale bar = 50 µm. BAL cytospins were immunostained for ATPβ (in blue), Ki-67 (in green), CD61 (in red) and angiostatin (in magenta). Representative images are shown in the lower image panels at A) 0, B) 24 and C) 36 h after O3 and LPS exposures. Scale bar = 50 µm. Please click here to view a larger version of this figure.
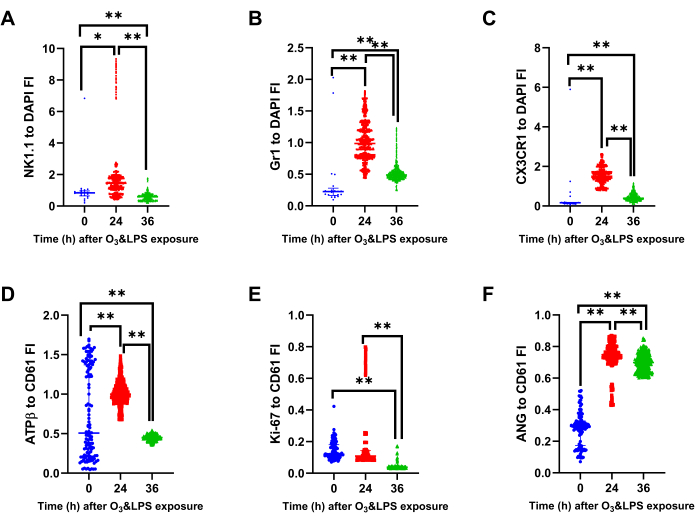
Figure 10: Bronchoalveolar lavage (BAL) mitochondrial protein ATPβ, lysosomal protein Gr1, intracellular CX3CR1 and angiostatin analysis: Immunostained BAL cells were normalized for DAPI to compute A) NK1.1 to DAPI fluorescent intensity (FI) ratio, B) Gr1 to DAPI FI ratio and C) CX3CR1 to DAPI FI ratio, at 0, 24 and 36 h after O3 and LPS exposures. Immunostained BAL cells were normalized for CD61 to compute A) ATPβ to DAPI fluorescent intensity (FI) ratio B) Ki-67 to DAPI FI ratio and B) Angiostatin (ANG) to DAPI FI ratio, at 0, 24 and 36 h after O3 and LPS exposures. For the graphs A-F, 0 h (n=21 cells) is represented in blue, 24 h (n=796 cells) in red and 36 h (n=2692 cells) in green data points. * represents p<0.05, ** represents p<0.01. Please click here to view a larger version of this figure.
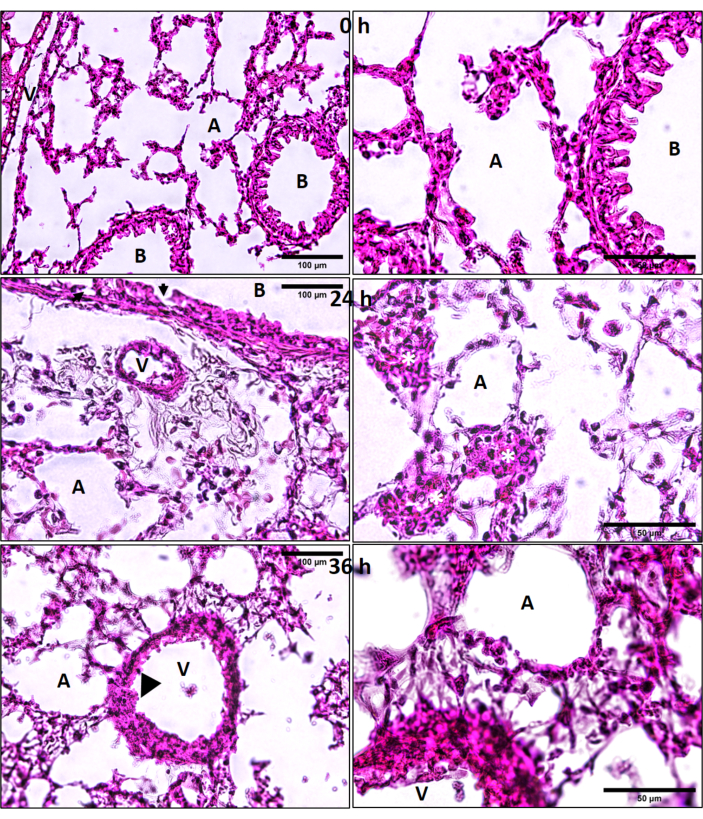
Figure 11: Lung hematoxylin and eosin (H&E) histology. Ozone (O3) and LPS induced lung cryosection H&E histology at 0, 24 and 36 h after combined O3 and LPS exposures. Regions of alveolar epithelial damage are marked by solid black arrows and endothelial damage by black arrow heads. Yellow asterisks (*) represent patches of leukocytes in the alveolar septal regions. A = alveolar space, B = bronchus, V = vasculature, scale bar = 100 µm for left hand image panels and 50 µm for image panels on the right side. Please click here to view a larger version of this figure.
| S.No. | Primary Antibody | Invitrogen Secondary Antibody |
| 1 | Mouse anti-NK1.1 IgG2a kappa (clone PK136), Invitrogen Catalog No. 16-5941-82 | Alexa 488 conjugated goat anti-mouse IgG (H+L), Catalog No. A11002 |
| 2 | Rat anti-Ly6G/Ly6C (Gr1) IgG2b kappa (clone RB6-8C5), Invitrogen Catalog No. 53-5931-82 | Alexa 568 conjugated goat anti-rat IgG (H+L), Catalog No. A11077 |
| 3 | Rabbit anti-CX3CR1 IgG (RRID 467880), Invitrogen Catalog No. 14-6093-81 | Alexa 633 conjugated goat anti-rabbit IgG (H+L), Catalog No. A21070 |
| 4 | Mouse anti-ATP5A1 IgG2b (clone 7H10BD4F9), Invitrogen Catalog No. 459240 | Alexa 488 conjugated goat anti-mouse IgG (H+L), Catalog No. A11002 |
| 5 | Rat anti-Ly6G IgG2a kappa (clone 1A8), Invitrogen Catalog No. 16-9668-82 | Alexa 568 conjugated goat anti-rat IgG (H+L), Catalog No. A11077 |
| 6 | Mouse anti-ATP5β IgG2b (clone 3D5AB1), Thermofisher Catalog No. A-21351 | Alexa 350 conjugated goat anti-mouse IgG (H+L), Catalog No. A11045 |
| 7 | Rat anti-Ki-67 (clone SolA15) IgG2a kappa, Invitrogen Catalog No. 14-5698-82 | Alexa 568 conjugated goat anti-rat IgG (H+L), Catalog No. A11077 |
| 8 | Armenian hamster anti-CD61 (clone 2C9.G2) IgG1 kappa, BD Catalog No. 553343 | Alexa 568 conjugated goat anti-hamster IgG (H+L), Catalog No. A21112 |
| 9 | Rabbit anti-angiostatin (mouse aa 98-116) IgG, Abcam Catalog No. ab2904 | Alexa 633 conjugated goat anti-rabbit IgG (H+L), Catalog No. A21070 |
Table 1: Primary and secondary antibody combinations used to stain cytospins prepared from the samples.
| Read-out | BAL | LVP | PB | SBM | FBM |
| TLC | * | * | + at 24 h; – at 36, 72 h | + at 72 h | |
| Neutrophils | + at 24, 36, 72 h | + at 24 h | * | + at 4, 24, 36, 72 h | + at 4, 24, 36, 72 h |
| Protein | + at 36 h | * | n.d. | n.d. | n.d. |
| Chemokine profile | – For eotaxin-2, IL-2 at 4, 24, 36, 72 h | + for Eotaxin-1/2, IL-2, TNFα, IFNγ, CX3CL1, CCL27, IL-16, CXCL5, CXCL10, MIP3β at 4 h; + for SDF1α at 36 h | n.d. | n.d. | n.d. |
| Cell size | + at 4 h; – at 24, 36 h | n.d. | n.d. | n.d. | n.d. |
| ATPα | – at 24 h | n.d. | n.d. | n.d. | n.d. |
| NK1.1/Ki-67 | + at 24 h | n.d. | n.d. | n.d. | n.d. |
| ATPβ/Gr1/CX3CR1/ANG | + at 24, 36 h | n.d. | n.d. | n.d. | n.d. |
Table 2: A comprehensive summary of the main findings of the study in different compartments. * denotes no change, + denotes an increase and – denotes a decrease in the measured parameters. BAL denotes broncho-alveolar lavage fluid, LVP denotes lung vascular perfusate, PB denotes peripheral blood, SBM denotes sternum bone marrow and FBM denotes femur bone marrow compartment.
Supplementary Figure 1: Gr1 and CD11b immunostaining: Representative images from DAPI (in blue), Gr1 (in green) and CD11b (in red) fluorescent immunostained cytospin slides of lung vascular perfusate (LVP), sternal bone marrow (BM) and femur bone marrow (BM) compartments at 0, 4 and 24 h after combined O3 and LPS exposures. Scale bar = 50 µm. Please click here to download this File.
Supplementary Movie 1 Three-dimensional rendered volume of BAL macrophages from 0 h time-point (i.e., baseline), showing nucleus (shown in blue) and immunostained for ATPα (shown in green) and Ly6G (shown in red). Please click here to download this Movie.
Supplementary Movie 2: Three-dimensional rendered volume of BAL macrophages from 24 h time-point after combined O3 and LPS exposures, showing nucleus (shown in blue) and immunostained for ATPα (shown in green) and Ly6G (shown in red). Note the presence of anuclear ATPα positive bodies as well as a fragmented cell. Please click here to download this Movie.
