Tumor immune infiltration is a parameter of the host anti-tumor response. Tumors are heterogeneous in the composition, density, location, and functional state of infiltrating leukocytes which interactions with cancer cells can underlie clinically relevant information to predict disease course and response to therapy. In this sense, microfluidic technologies could be used as complementary and privileged in vitro tools to explore the immune contexture of tumors, as well as to monitor the response to anticancer therapies. The coupling of the microfluidic assay, live-cell imaging, and tracking software may establish reliable quantification methods to quantify how immune cells adjust their migration pattern in different contexts. In this chapter, we have reported steps for setting up versatile 2D or 3D co-cultures of immune and target cancer cells in ad hoc microfluidic devices realized by standard soft-lithographic procedures. In Section 1 microfluidic devices were employed to allow chemical and physical contacts between adherent (MDA-MB-231 cancer cells) and non-adherent (PBMCs) populations. Some chemotherapeutic agents (e.g., anthracyclines among the others) can induce "immunogenic" apoptosis of malignant cells, thus enhance their visibility in immunocompetent hosts. Cancer immunogenic cell death (ICD) is characterized by the release of membrane-bound and soluble signals delivered by dying cells functioning as alarmins for immune cells. To provide a quantitative validation of ICD response, we employ data collected in the microfluidic platform where leukocytes can move through suitably built microchannel bridges toward their target cells. Time-lapse recordings were performed after co-loading PBMCs, from healthy donors (WT, wild type), with human MDA-MB-231 breast cancer cells pre-treated or not with the anthracycline DOXO. Microphotographs were generated every 2 min, for two successive time intervals of 24 and 48 h. (Exemplary Movie S1 of 0-24 h interval). Tracking analysis of individual PBMCs challenged with dying (DOXO-treated) or live (PBS-treated) cancer cells was done using Trackmate plug-in, as seen in Figure 6A (left panel). Relevant chemotaxis values and migration plots were automatically generated by using the Chemotaxis and Migration Tool, as detailed in62. The cell trajectories were all extrapolated to (x, y) = 0 at time 24 h. The results indicated a different migratory profile of the immune cells when co-loaded with breast cancer cells exposed to DOXO or PBS. When PBMCs were confronted with apoptotic cancer cells, they crossed microchannels towards dying/dead cells (but not to live untreated cells). Migration X/Y spider and rose plots, presented in left panel Figure 6B-C, were mapped and compared to highlight the impact of immunogenic inducers agents on differences in immune dynamics. Rose plots demonstrate that PBMCs migrated mainly in nearly all directions in the control experiment, only a negligible fraction is guided up the gradient generated by proliferating cancer cells. Conversely, the paths of individual cells highlight a strong bias movement along the direction of apoptotic breast cancer cells (negative x-direction). To evaluate directed immune cell migration towards DOXO-treated or PBS-treated cancer cells, several chemotaxis parameters are computed, including: a) the center of mass (spatial averaged point of all endpoints); b) the directionality; c) the forward migration index (i.e., the average cell displacement in a direction of interest which means towards target tumor site, in our case). The latter values represent a measurement of the efficiency of a cell to migrate in the direction given chemotactic stimuli. After reaching the left chamber, a fraction of leukocytes with an increasing density over 24-48 h exhibited long-term (>60 min) contacts with DOXO-treated MDA-MB-231. PBMCs fail to massively migrate and engage in such long-term and persistent interactions with live cancer cells (as shown by representative microphotographs of approaching PBMCs in Figure 6A, right). For quantifying differences in tumor-immune interplay, the tumor region was delineated with a fixed circle of diameter ranging 20-80 microns, named "hotspot". The quantification and analysis from representative FOVs are shown in Figure 6 (Right panel, B-C).
In Section 2 a novel 3D immuno-competent tumor model was described to quantify the recruitment of immune cells in response to anti-cancer combinations of epigenetic drugs32 (DAC/IFN vs IFN alone), allowing the comparison of two different treatment conditions of A375 melanoma cells simultaneously. Thus, A375M melanoma cells, labeled with PKH67 green fluorescent dye were embedded in Matrigel matrices in the presence of DAC and/or IFN into each gel chamber, whereas PKH26 red-labeled PBMCs were distributed homogenously into the central fluidic chamber at the starting point (Figure 7A). We compared simultaneously the two tumor masses in 3D matrices for their capacity to attract PBMCs. At 48 and 72 h, PBMCs are guided massively into the right-side microchannels, as observed in Figure 7B.
A preferential homing of PBMCs toward the gel matrix containing DAC/IFN-treated, rather than IFN-treated, melanoma site was clearly observed, whereas poor migration rate was visible toward A375 cells exposed to single treatment (left chip side). The competitive setting is applicable to investigate a plethora of different cancer biology phenotypes (e.g., drug-resistant vs aggressive, primary or metastatic (e.g., A375P vs A375M melanoma cells), and responders vs non-responders). Gel chambers can be comprised of complex co-cultures of malignant cells and multiple non-cancerous tumor-associated cells, such as endothelial cells, immune cells, and fibroblasts33 to characterize TME-level drug responses. Events of mitosis and apoptotic death of cancer cells can be monitored by staining with live dyes33. Immunostaining procedures on 3D microfluidic devices could be adapted to evaluate states of activation of infiltrated immune cells in tumor sites by confocal microscopy35. In this sense, these 3D microfluidic systems may mimic complex tumor structures and multicellular interactions and therefore are valuable platforms for more reliable preclinical drug testing.
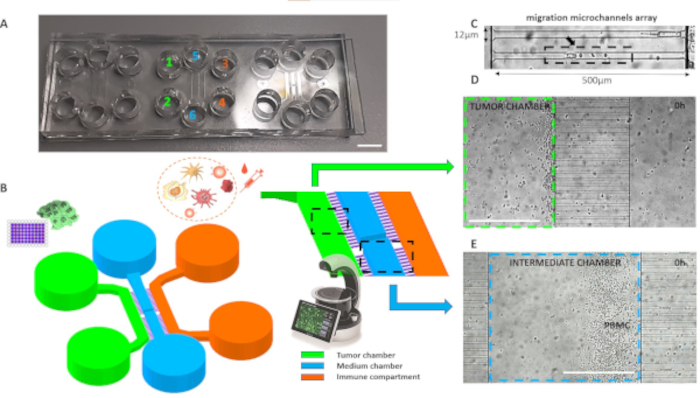
Figure 1. Planimetry of the microfluidic device for assembling 2D tumor-immune co-cultures. (A) Real microphotograph of 3 chips assembled into a single microscope slide. The reservoirs are numbered depending on the loading sequence and are color-coded as the corresponding culture chambers listed in the legend. Scalebar, 6 mm. (B) 3D CAD of chip consisting of three main cell culture areas connected by microgrooves. C) Details of the narrow microgrooves bridge, showing the dimensions tailored to cancer cells and PBMCs size. D-E) Visible light microphotograph was acquired before the beginning of the time-lapse, showing the distribution of cancer cells and PBMCs respectively in the left and the intermediate chamber. Scalebar, 500 µm. Please click here to view a larger version of this figure.
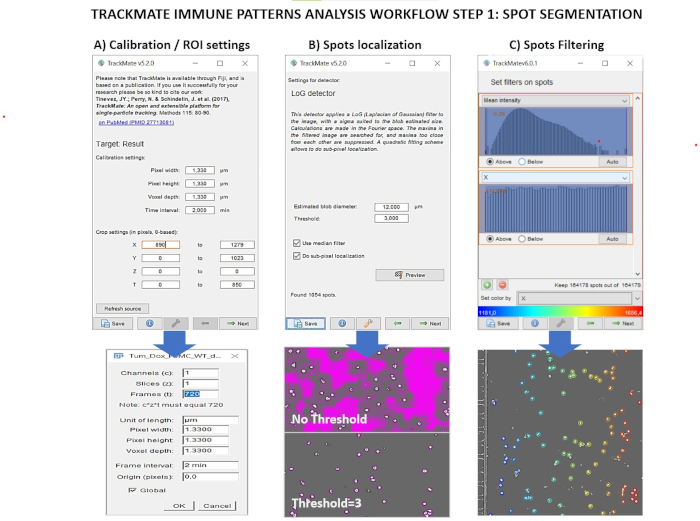
Figure 2. Trackmate analysis pipeline for localization of immune spots in time-series images. A) Upper panel: Screenshot of Trackmate image calibration menu. Lower panel: Fiji "Image properties menu" to set time and spatial units. B) Upper panel: Screenshot of Trackmate "Spots detection" menu. Lower panel: Output image with applied different values of "Threshold". C) Upper panel: Screenshot of Trackmate "Spot filtering" menu illustrating some filters. Lower panel: Exemplary time-lapse image, depicting immune spots filtered by position along X in the intermediate chamber. Please click here to view a larger version of this figure.
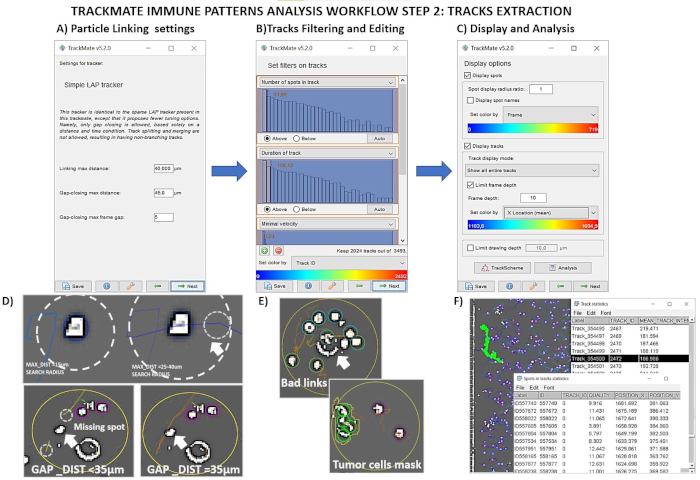
Figure 3. Trackmate Analysis pipeline for reconstructing immune tracks in time-series image sequences. A-C) Screenshots of Trackmate menus for tracks building, tracks filtering, and for exporting final data. D) Examples of generated tracks in pre-processed time-lapse images varying the linking detector settings. E) Example of false tracks and bad links derived from the detection of tumor cells borders. F) Tracks are highlighted in green by selecting rows in the file .txt of results. Please click here to view a larger version of this figure.
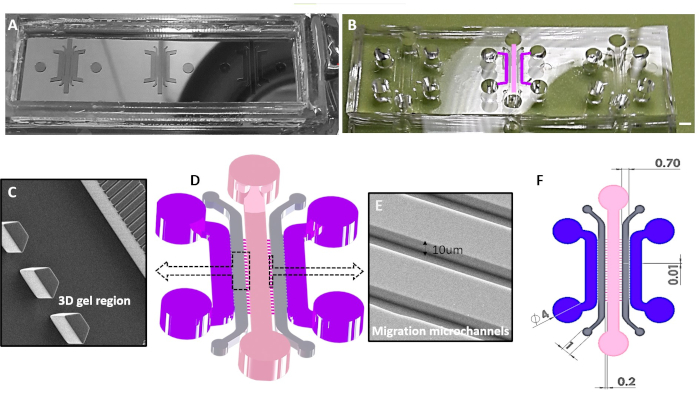
Figure 4. Schematic overview for 3D immunocompetent tumor-on chips. A) Example of a micro-structured silicon master. Stamps for PDMS were patterned in SU-8 negative resist in a cleanroom facility equipped with e-beam and optical lithography. B) PDMS replicas were fabricated by standard soft-lithography methods. The central unit has the chambers colored as the ones drawn in 3D rendering of the chip, depicted in the panel D. C) Scanning electron microscopy (SEM) enlarged view of the array of micropillars suited for hydrogel solution entrapment. D)3D CAD showing the chambers, connected by microgrooves and the loading wells. Dashed boxes are referred to SEM photographs of details (panel C, and E). E) SEM photograph of 10 µm high connecting microgrooves. F) 2D CAD layout depicting the dimensions of microstructures. Please click here to view a larger version of this figure.
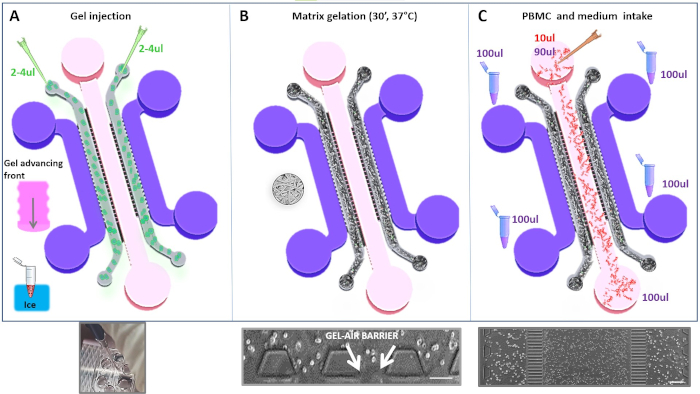
Figure 5. Schematic workflow of main loading protocol steps for assembling 3D co-cultures. A) Upper panel: Schematics of injection step of Matrigel solution in each gel side region. Lower panel: Real photograph of the chip showing gel front advancement along the channel during loading step. B) Schematics of Matrigel polymerization step. Lower panel: Phase-contrast microscopic view of the gel-air interface formed after gelation step. Scalebar, 100 µm. C)Upper panel: Drawing, depicting the loading of cells and media with exemplary volumes used in the protocol. Lower panel: A 4X Phase-contrast image of the assembled co-culture at 0h is shown. Scalebar, 200 µm. Please click here to view a larger version of this figure.
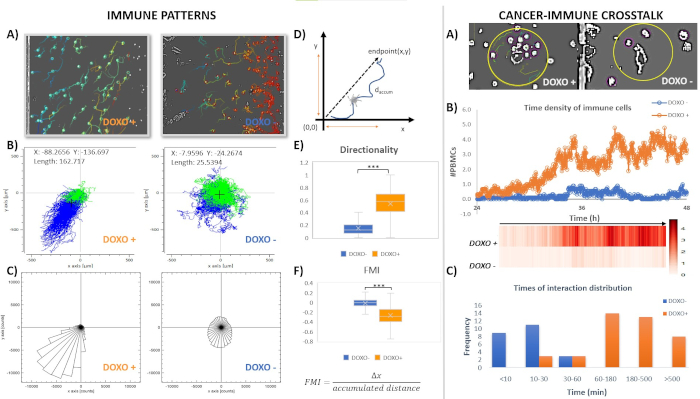
Figure 6. Analysis of migratory profiles and interaction behavior of PBMC towards dying/live tumor chamber in the time window of 24-48 h. Left panel. A) Screenshots of time-lapse pre-processed images overlaid with immune colored tracks, extracted by Trackmate. The immune paths are obtained by a single FOV in the indicated experimental conditions in the interval 24-to-48 h in the intermediate chamber. B-C) Representative migration tracks and rose plots of PBMCs cultivated in the two different conditions (n = 1550 PBMCs versus DOXO-treated cancer cells, DOXO+ or n =1434 PBMCs versus control cancer cells, DOXO-). Each line inside x-y plots depicts a single PBMC trajectory and each circle represents the final position of a single cell with respect to the initial position. Starting points are set to (0,0) using a coordinate transformation. The coordinates and displacement of the center of mass of cell migration are displayed in the indicated experimental conditions. The center of mass coordinates and displacement (computed as average Euclidean distance for the X and Y components) provide indication of the average direction in which the group of cells primarily travelled and magnitude of the overall cell movement in condition groups. Blue and green lines respectively mark individual tracks by Euclidean distance value greater/smaller than a threshold value (100 µm). D) Scheme of numerical data extracted by a cell track. E-F) Box & Whiskers plot depicting respectively directionality (p<0,0001 Unpaired t test with Welch's correction) and FMI (p<0,0001 Unpaired t test with Welch's correction). The horizontal line in boxes represents median values. The FMI values are the average for all cell-tracks in each condition group.Right panel. A) Screenshots of Trackmate extracted ROIs showing differential interactions between PBMCs and DOXO or PBS-treated cancer cells. B) Time density of PBMCs around DOXO-treated or control MDA-MB-231 cancer cells. Number of PBMCs present into selected ROI around cancer cells for each condition. Values reported are the average over 9 selected ROI from a single time-lapse FOV. Cancer hotspots are defined in a ROI (80 µm in diameter). Corresponding density heatmap is displayed. Dots represent mean number of cells calculated over 9 cancer hotspots at indicated time points. C) Distribution of times (min) of contacts for each experimental group. Please click here to view a larger version of this figure.
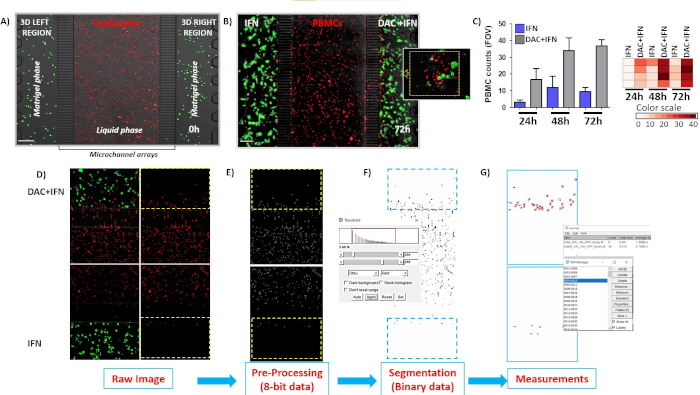
Figure 7. Preferential recruitment of PBMCs in response to DAC plus IFN drug combinations in a competitive 3D immuno-competent melanoma on chip model. A) Distribution of initially loaded PBMCs in the central chamber of microfluidic devices. Microphotographs are acquired by EVOS-FL fluorescence microscope during 0-72 h interval. Red fluorescence (PKH67-labeled cells) represents PBMCs from healthy donors. PKH67-labeled (green) human melanoma cells embedded in Matrigel containing single or double combinations treatments were plated in lateral chambers. B) A375 cells plus IFN on left versus A375 plus DAC/IFN on the right. Fluorescence images were shown at 72 h of co-culture. Discontinued yellow box depicts visibly massive recruitment in A375 plus DAC/IFN side. C) PBMC counts in four different ROIs from IFN chambers vs DAC+IFN chambers. Histograms represent cell counts +/- S.D.; heatmap enumerated values from each ROI. Scalebars, 200 µm. D-G) Schematics of segmentation and quantification steps of infiltrated PBMCs in gel matrices applied to single channel fluorescence images. Please click here to view a larger version of this figure.
Supplementary File. Microfabrication protocol Please click here to download this file.
Supplementary Movie 1. Time-lapses sequences in a 2D tumor-immune on-chip co-culture. Microphotographs were acquired every 2 min, in a time interval of 0-24 h by means of a compact microscope placed into a standard cell culture incubator. Left panel. Movie of PBMCs from healthy donors (WT, wild type), plated with MDA-MB-231 control breast cancer cells. Right panel. Movie of a massive migration of PBMCs WT towards the chip chamber where MDA-MB-231 DOXO-treated cancer cells, are loaded. 2D chip layout is described in detail in section 1of Protocol and shown in Figure 1. Please click here to download this movie.
