Confocal images of larval and adult optic lobes mounted in the orientations described in the protocol are presented in Figure 1 and Figure 2.
Figure 1 shows schematics and representative confocal slices of larval brains positioned in the anterior, posterior and lateral orientations. In the anterior mounting orientation, the OPC epithelium (DE-Cadherin), medulla neuroblasts (deadpan>βgal) and lamina neurons (Dachshund) appear at the surface as bands of cells that wrap around the brain (Figures 1B,D). The OPC is spatially patterned along its dorsal/ventral axis by the differential expression of transcription factors6,11,12. Vsx1 labels the central OPC (cOPC), Optix labels the main OPC (mOPC) and Rx is expressed in the posterior OPC (pOPC), which represents the tips of the crescent11,12,25. The anterior mount is ideal for the visualization of the cOPC, as this region of the crescent lies at the surface in this orientation (Figure 1D). Neurons of the lamina are also visible on the lateral side of the lobe in this orientation. Deeper into the brain along the z-axis, additional regions of the OPC, as well as other structures, become visible (Figure 1E,F). At a middle point in the z-stack, the mOPC epithelium, along with its respective neuroblasts and neurons, are visible (Figure 1E). Additionally, the proximal region of the IPC (p-IPC) and the lobula plug, which give rise to neurons of the lobula and lobula plate6,13,26, are visible in these intermediate slices. The deepest z-slices depict the other side of the brain, where the pOPC tips are located (Figure 1F). The superficial tip of the IPC (s-IPC) is also present in these deepest slices13.
The structures and cells located at the bottom of an anteriorly-mounted brain correspond to those that would appear on the surface of a posteriorly-mounted brain. Due to their proximity to the imaging objective, the optic lobe structures closest to the surface of the brain resolve better in confocal images compared to those located at the bottom. At the surface, there is minimal light scattering between the tissue and the objective. In deeper parts of the tissue, more light scattering leads to a weaker fluorescence signal. Thus, a posterior mounting strategy permits optimal imaging conditions to visualize the tips of the OPC or ventral-IPC (Figure 1F), whereas an anterior mounting strategy is better-suited for visualizing cells of the lamina or cOPC (Figure 1D). If the region of interest is the lobula plug or mOPC and its progeny, either mounting orientation is suitable, as these structures are located towards the middle of the brain (Figure 1E).
A laterally-mounted larval brain lobe (Figure 1G) can be used to visualize the medulla, lamina or lobula plug neuronal crescents in one focal plane. Different structures can be visualized at different depths along the z-axis. At the surface, the lamina crescent (Eya) is visible with the lobula plug crescent located between its arms (Figure 1H). The medulla neuronal crescent (Bsh, Eya and Svp) appears at a slightly deeper z-position (Figure 1I). In a single z-slice one can visualize the entirety of the neuronal crescent along both the dorsal-ventral and anterior-posterior axes. Thus, this orientation is suitable for a researcher interested in determining where their gene of interest is expressed with respect to the spatial axes of the OPC.
Figure 2 shows schematics and representative confocal slices of adult brains positioned in the anterior, posterior and horizontal orientations. The lamina has been removed in these images to better show the underlying medulla and lobula complex. The medulla, lobula and lobula plate are each comprised of a cortex, which contains neuronal cell bodies, and a neuropil, which is made up of axonal and dendritic arborizations. In the anterior orientation (Figure 2B), the medulla cortex and neuropil are located at the surface, whereas in the posterior orientation (Figure 2C), the lobula and lobula plate are the first structures imaged. Figure 2D‒F display representative images of an anteriorly-mounted adult optic lobe at three Z-positions. Since the medulla is located at the surface of the anterior mount (Figure 2D), the medulla cortex (Vsx1) is immediately visible. Cell bodies in the cortex project their arborizations into the neuropil (labeled by Bruchpilot), which can be visualized at an intermediate z-position (Figure 2E). The lobula also appears at this level, located perpendicular to the medulla. At the deepest z-position, the lobula plate is visible (Figure 2F). Thus, researchers interested in studying the neurons of the lobula complex should use a posterior-mount orientation, whereas those interested in the lamina and medulla should mount their brains in an anterior orientation.
A horizontally-mounted optic lobe is achieved when the brain is flipped 90° on to its side from an initial anterior position (Figure 2G). In this view, all of the neuropils and cortices of the optic lobe are visible within a single plane (Figure 2H,I). This mounting orientation is recommended for the visualization of retinotopic projections and the projections of neurons that target multiple optic lobe neuropils.
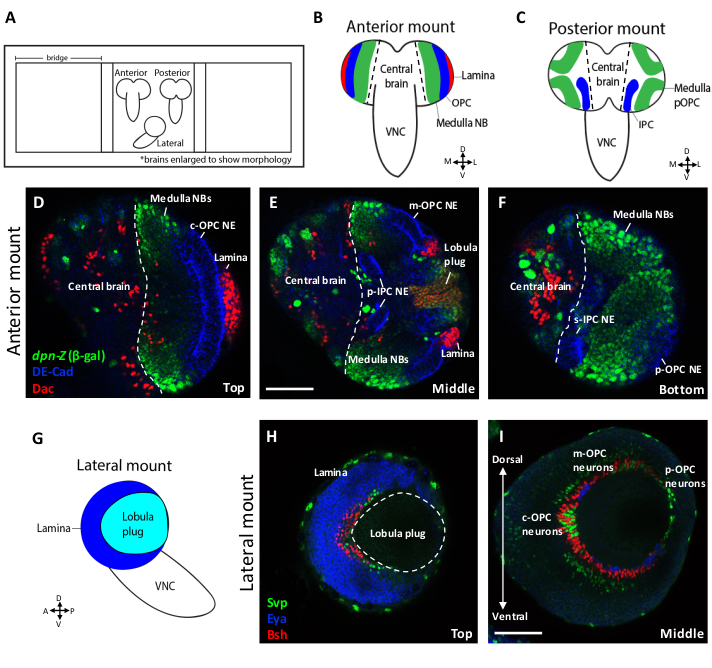
Figure 1. Larval brain mounting orientations. (A) Cartoon schematic showing late 3rd instar larval brains mounted on a slide in three orientations. Orientations can be distinguished from one another based on the position of the ventral nerve cord (VNC) relative to the brain lobes. (B) In the anterior mount, the VNC comes over the top of the brain lobes. In this orientation, the medulla neuroblasts (NBs), anterior OPC neuroepithelium (NE) and neurons of the lamina are visible. (C) In the posterior mount, the VNC protrudes from beneath the brain lobes. This orientation permits the visualization of both posterior (tip) medulla NBs and the ventral IPC NE. (D‒F) Confocal images of an anteriorly-mounted 3rd instar larval brain stained for the OPC and IPC NE marker DE-Cadherin (blue), lamina marker Dac (red) and NB marker dpn>lacZ (β-gal) (green). D-F are Z-slices from the top (D), middle (E) and bottom (F) of the same confocal stack. (G) Schematic depicting a late 3rd instar larval brain mounted in a lateral orientation. (H, I) Confocal images of a laterally-mounted 3rd instar larval optic lobe stained for the lamina marker Eya (blue), and lamina and medulla neuronal markers Bsh (red) and Svp (green). In this orientation, the lamina (H) and medulla (I) neuronal crescents can be visualized along the entire dorsal-ventral and anterior-posterior axes in a single plane. H and I are Z-slices from top (H) and middle (I) of the same confocal stack. Scale bars = 50 um. Please click here to view a larger version of this figure.
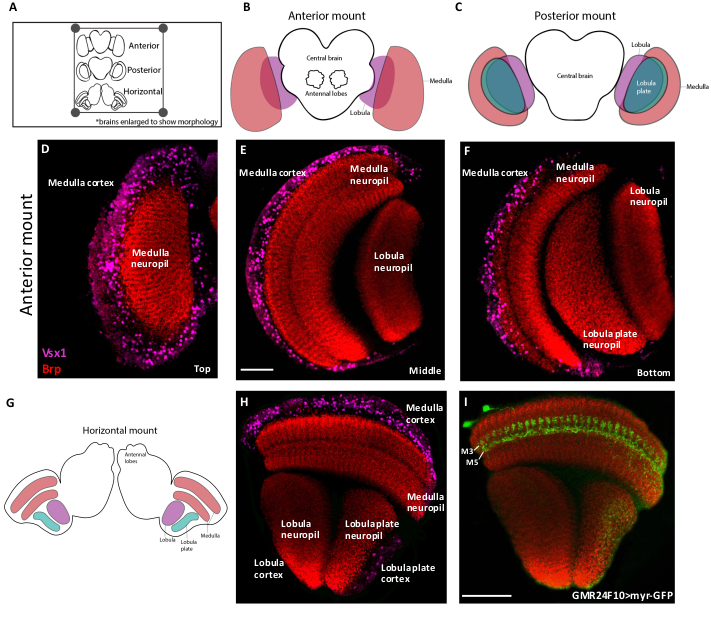
Figure 2. Adult brain mounting orientations. (A) Cartoon schematic showing adult brains mounted on a slide in three orientations. Orientations can be distinguished based on the location of the antennal lobes. (B) In the anterior orientation, the antennal lobes are facing up and the medulla neuropil is located at the surface. (C) In the posterior orientation, the brain is mounted with the antennal lobes facing down and the lobula and lobula plate neuropils located at the surface. (D‒F) Confocal Z-slices of an anteriorly-mounted adult brain labeled with the medulla neuronal marker Vsx1 (magenta) and the neuropil marker Brp (red). D-F are Z-slices from the top (D), middle (E) or bottom (F) of the same confocal stack. (G) Schematic of an adult brain in the horizontal orientation, which can be achieved by mounting the brain on its side. (H) Confocal image of a horizontally-mounted brain stained with Vsx1 (magenta) and Brp (red). In this mounting orientation all three optic lobe neuropils and cortices are visible in one Z-slice. This orientation is ideal for visualizing the morphologies of optic lobe neurons across the neuropils within a single plane. (I) Confocal image of a horizontally-mounted brain labeling a medulla neuron (Dm4) with GFP (green) and the neuropils with Brp (red). Dm4 neurons send arborizations to layers 3 and 5 of the medulla neuropil. Scale bars = 50 µm. Please click here to view a larger version of this figure.
