Isolation of Specific Neuron Populations from Roundworm Caenorhabditis elegans
Summary
Here, we present a protocol for simple isolation of specific groups of live neuronal cells expressing green fluorescent protein from transgenic Caenorhabditis elegans lines. This method enables a variety of ex vivo studies focused on specific neurons and has the capacity to isolate cells for further short-term culturing.
Abstract
During the aging process, many cells accumulate high levels of damage leading to cellular dysfunction, which underlies many geriatric and pathological conditions. Post-mitotic neurons represent a major cell type affected by aging. Although multiple mammalian models of neuronal aging exist, they are challenging and expensive to establish. The roundworm Caenorhabditis elegans is a powerful model to study neuronal aging, as these animals have short lifespan, an available robust genetic toolbox, and well-cataloged nervous system. The method presented herein allows for seamless isolation of specific cells based on the expression of a transgenic green fluorescent protein (GFP). Transgenic animal lines expressing GFP under distinct, cell type-specific promoters are digested to remove the outer cuticle and gently mechanically disrupted to produce slurry containing various cell types. The cells of interest are then separated from non-target cells through fluorescence-activated cell sorting, or by anti-GFP-coupled magnetic beads. The isolated cells can then be cultured for a limited time or immediately used for cell-specific ex vivo analyses such as transcriptional analysis by real time quantitative PCR. Thus, this protocol allows for rapid and robust analysis of cell-specific responses within different neuronal populations in C. elegans.
Introduction
Over the past several decades, the metazoan model organism Caenorhabditis elegans has been a tremendous asset in the study of neurons, neuronal circuitry and its role in physiological and behavioral responses, and aging-associated neurodegenerative diseases. A unique feature of C. elegans is that the animals are transparent, allowing for the lineage of all adult somatic cells to be mapped1. C. elegans also harbors manageable quantity of neurons, leading to the morphology and connectivity of the nervous system being well understood2. The invariant cell lineage, short lifespan, and abundance of high-throughput genetic tools available for C. elegans make it an ideal model organism for the study of aging within different neuronal populations.
Aging of neurons and neurodegenerative disorders are complex, and each disorder has unique pathological signatures associated with it. However, for many of these disorders, such as Parkinson’s and Alzheimer’s disease, a common feature is the progressive load of misfolded proteins3,4,5. In both of these diseases, proteins misfold and aggregate within the cell to cause toxicity, ultimately leading to cell death4,5. For proper aging of neurons, the homeostasis of mitochondria, more specifically protein homeostasis, is important, as perturbations and dyshomeostasis can lead to neurodegeneration6,7,8. Cells are equipped with various mechanisms to maintain protein homeostasis, one being the unfolded protein response (UPR)―a classical pathway where signals from endoplasmic reticulum (ER) activate intracellular signal cascades, leading to a transcriptional response9. Akin to this UPRER, metazoans display a similar response to loss of mitochondrial protein homeostasis, the so-called mitochondrial UPR (UPRmt)7,8. C. elegans appears to be the most basal organism to have a confirmed and well-defined UPRmt pathway7.
The function/dysfunction of specific neuronal populations within a larger network can be difficult to assess due to their intrinsic connectivity and complexity3. However, there is often a need to study distinct neuronal populations due to cell type-specific diseases with complex pathologies, such as those associated with impaired cellular protein homeostasis. In C. elegans, specific neuronal populations can be genetically manipulated allowing for facile observation in vivo. The nervous system of C. elegans primarily consists of neurons, with a small percentage of glial cells. In adult hermaphrodite worms, there are approximately 302 neurons, subdivided into over 100 different classes2,10. Neurons such as cholinergic neurons predominate in neuromuscular junctions, while dopaminergic neurons are primarily involved in sensation10,11. As both motor activity and sensory abilities decline with age, it is important to detail mechanistic insights about defects in these individual neurons11,12. As such, there is a need for a simple and robust method to isolate intact cells of interest for subsequent ex vivo studies.
Here, we describe an optimized effective method for the rapid isolation of specific neuronal cells expressing green fluorescent protein (GFP) from C. elegans. This isolation method can be performed on larval, juvenile or adult worms. Isolation of cells from larval worms was previously published by Zhang et al.13 and will not be discussed here. An important note here is that all worms need to be at the same life stage to prevent over-digestion of the animal or contamination from animals in different life stages. Through both enzymatic and mechanical disruption of the nematode cuticle, an exoskeleton high in collagen and other structural proteins, a wide variety of cells can be isolated13,14. Cells can then be isolated via flow cytometry or antibody labeled magnetic beads. Typically, RNA is isolated via a phenol and guanidine isothiocyanate method to ensure the enrichment of desired cell population15. With much care these isolated cells can be maintained in a culture flask or multi-well dish. This method represents a unique and powerful tool in the study of specific neurons and has the capacity to isolate live and functional cells for further culturing.
Protocol
1. Preparation and collection of aged worms for cell isolation
NOTE: Explained below is the isolation of cholinergic neurons from the transgenic unc-17::GFP strain (OH13083) obtained from the Caenorhabditis Genetics Center (CGC) strain repository at the University of Minnesota. It is imperative to maintain sterile conditions to prevent contamination from fungi or bacteria.
- Prepare and synchronize worms via the bleaching method as described by T. Stiernagle16. For aging experiments, plate worms onto nematode growth medium (NGM) plates containing 25 µM water solution of fluorodeoxyuridine (FuDR) to reduce egg production and arrest egg hatching. Inspect agar plates regularly to prevent contamination or egg hatching as this will cause unreliable results.
NOTE: For unc-17::GFP cells, typically three 100 mm x 15 mm NGM plates are used to isolate a sufficient amount of cells of interest16. - Collect worms and prepare buffers for cell isolation.
- Collect worms in a 15 mL conical tube. Wash worms from plate with 1.5 mL of M9 buffer (1 mM MgSO4, 85 mM NaCl, 42 mM Na2HPO4·7H2O, 22 mM KH2PO, pH 7.0) and centrifuge for 5 min at 1,600 x g. Discard supernatant and wash worms with 1 mL of M9 buffer. Repeat centrifugation and wash for a total of five times to remove as much E. coli contamination as possible.
NOTE: Adding antibiotics (50 µg/mL), such as ampicillin, at this step can help reduce bacterial contamination. - For two samples, prepare 2 mL of sodium dodecyl sulfate-dithiothreitol (SDS-DTT) lysis buffer: 200 mM DTT, 0.25% (w/v) SDS, 20 mM HEPES (pH 8.0), and 3% (w/v) sucrose.
- Prepare 15 mL of isolation buffer (118 mM NaCl, 48 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 25 mM HEPES [pH 7.3]) and store it on ice.
NOTE: Both the SDS-DTT lysis buffer and isolation buffer must be made fresh before each experiment.
- Collect worms in a 15 mL conical tube. Wash worms from plate with 1.5 mL of M9 buffer (1 mM MgSO4, 85 mM NaCl, 42 mM Na2HPO4·7H2O, 22 mM KH2PO, pH 7.0) and centrifuge for 5 min at 1,600 x g. Discard supernatant and wash worms with 1 mL of M9 buffer. Repeat centrifugation and wash for a total of five times to remove as much E. coli contamination as possible.
- Cuticle disruption and single cell isolation
- Centrifuge animals collected in step 1.2.1 for 5 min at 1,600 x g. Remove all supernatant and suspend worms in 1 mL of M9 media and transfer to 1.5 mL microcentrifuge tube.
- Pellet worms with centrifugation at 1,600 x g for 5 min.
- Add 200 µL of SDS-DTT lysis buffer to worms and incubate for 5 min at room temperature (RT). Worms should appear to be “winkled” along the body if viewed under a light microscope.
NOTE: Prolonged exposure to SDS-DTT lysis buffer may result in death of worms, which can be monitored by observing worms. Worms that are dead will elongate and will not curl. - Add 800 µL of ice-cold isolation buffer and mix by gently flicking tube.
- Pellet worms for 1 min at 13,000 x g at 4 °C, remove supernatant and wash with 1 mL of isolation buffer.
- Repeat step 1.3.5 for a total of five times, carefully removing isolation buffer each time.
- Add 100 µL of protease mixture from Streptomyces griseus (15 mg/mL) (Table of Materials) dissolved in isolation buffer to the pellet and incubate for 10−15 min at RT.
NOTE: As with the SDS-DTT lysis buffer, the extended protease digestion may result in excessive cleavage of proteins along the plasma membrane, preventing isolation of surface exposed GFP via magnetic beads. - During incubation with protease mixture, apply mechanical disruption by pipetting samples up and down against the bottom of the 1.5 mL microcentrifuge tube with a 200 µL micropipette tip for ~60−70 times. Keep the pipette tip against the wall of the microcentrifuge tube with constant pressure to properly disassociate cells.
- To determine the stage of digestion, remove a small volume (~1−5 µL) of the digestion mixture, drop it on a glass slide and inspect it using a tissue culture microscope. After 5−7 min of incubation, worm fragments should have visibly reduced cuticle and a slurry of cells will be readily visible.
- Halt reaction with 900 μL of commercially available cold Leibovitz’s L-15 medium supplemented with 10% fetal bovine serum (FBS) and penicillin-streptomycin (final concentration at 50 U/mL penicillin and 50 μg/mL streptomycin).
- Pellet isolated fragments and cells by centrifugation for 5 min at 10,000 x g at 4 °C. Wash the pelleted cells with 1 mL of cold L-15-supplemented media twice more to ensure that excess debris and cuticle is removed.
- Resuspend pelleted cells in 1 mL of L-15-supplemented media and leave on ice for 30 min. Take the top layer, approximately 700−800 µL, to a microcentrifuge tube. This layer contains cells without cell debris and will be used in the subsequent isolation of cells of interest.
- Following manufacturer’s instructions, use an automated cell counter or hemocytometer to measure the cell density of 10−25 µL of isolated cells.
2. Isolation of GFP-positive cells via flow cytometry or anti-GFP magnetic beads
NOTE: There are two methods that can be deployed to isolate specific cell types. The first method is to use fluorescence-activated cell sorting (FACS), while the second method uses antibody-conjugated magnetic beads to pool cells expressing distinct plasma membrane-localized proteins. The latter method requires the localization of GFP to the plasma membrane, thus being available for interaction with the antibody-coupled magnetic bead.
- Isolation of GFP-positive cells by flow cytometry
- From isolated cell suspension collected in step 1.3.12, centrifuge cells for 5 min at 10,000 x g at 4 °C. Discard supernatant and suspend pelleted cells in L-15-supplemented medium to a cell density of no more than 6 x 106 cells/mL to avoid overloading the flow cytometer.
- Sort cells by GFP-positive expression using a flow cytometer capable of sorting samples. GFP-negative cells can be used as a control for non-cell type specific analysis.
NOTE: As mentioned above, an alternative approach to isolate GFP-positive cells may be used if the GFP-tagged protein of interest is localized to the cells’ plasma membrane. In this case, the protocol described in section 2.2 can be employed.
- Isolation of GFP-positive cells via anti-GFP magnetic beads
- From isolated cell suspension collected in step 1.3.12, centrifuge cells for 5 min at 10,000 x g at 4 °C. Discard supernatant and suspend cells in 485 µL of L-15-supplemented medium. Add 15 µL of ddH2O pre-washed α-GFP antibody-coupled magnetic beads to the solution.
- Incubate cells with the magnetic bead slurry at 4 °C for 1 h with very gentle turning. Excessive turning will damage the cells.
- After incubation, centrifuge sample for 5 min at 10,000 x g at 4 °C, then wash pellet 2x with 1 mL of L-15-supplemented medium to remove GFP-negative cells.
- Culturing of isolated cells
- Plate isolated cells into a sterile 6-well multi-well plate.
NOTE: These plates may be coated with peanut lectin to increase cell attachment; however, if cells are to be used immediately, this step may be disregarded. - Place the plate into a plastic container and incubate cells at 20 °C with a damp wipe or soft tissue paper (add 50 µg/mL ampicillin to ddH2O to ensure no bacterial growth on wipe) to add moisture to cells. Do not incubate in a CO2 incubator, as elevated CO2 levels may damage the cells.
- Plate isolated cells into a sterile 6-well multi-well plate.
3. Fluorescent imaging of isolated GFP-positive cells
- Turn on the fluorescent bulb of an inverted microscope with fluorescence attachment and allow it to warm up for about 10 min.
- Remove cell culture from the incubator and set on stage of inverted microscope with florescence attachment. Slide the GFP filter into the grooves under the stage of the microscope.
- View the cells with a magnification of 50x. Successfully isolated GFP-positive cells will be readily visible. Take photos using the camera attachment on the microscope.
4. Extraction of RNA from isolated cells
NOTE: Extraction of RNA should be performed immediately after cell isolation to ensure high quality of material. Isolated RNA can be used to produce cDNA and subsequent measurement of transcript levels by quantitative PCR (qPCR). All tubes, tips, and reagents should be RNase-free, and workspace should be ethanol-washed prior to RNA extraction.
- From isolated cells collected at step 2.1.2, centrifuge cells in a 1.5 mL sterile, RNase-free microcentrifuge tubefor 5 min at 10,000 x g at 4 °C. If cells are isolated via magnetic beads, follow cell collection as stated above.
- Using a micropipette, remove supernatant from centrifuged microcentrifuge tube and add 100 µL of M9 medium to wash away L-15-supplemented medium. Centrifuge cells 5 min at 10,000 x g at 4 °C and remove supernatant. To ensure all media are removed, repeat for a total of three washes carefully removing supernatant each time.
- Add 1 mL of phenol and guanidine isothiocyanate solution (Table of Materials) to cells and pipette the suspension up and down carefully. Incubate the mixture at RT for 5 min.
- Following incubation, add 250 phenol and guanidine isothiocyanate solution
- Centrifuge the solution for 5 min at 10,000 x g at 25 °C.
- Carefully remove as much of the top aqueous layer with a micropipette as possible, without disturbing the bottom organic layers, and transfer the bottom layer to a new 1.5 mL RNase-free microcentrifuge tube. To the new tube, add 500 µL of isopropanol and mix by inverting the tube. Incubate this solution at RT for 5 min.
- Centrifuge the tube at 14,000 x g for 20 min at 25 °C.
- While keeping the samples on ice, remove as much isopropanol as possible with a micropipette, and gently add 1 mL of 70% (v/v) ethanol in RNase-free ddH2O to the tube. Be sure not to eject solution directly onto the bottom of the tube; apply ethanol/water mixture to the side of the tube.
- Centrifuge at 10,000 x g for 5 min at 25 °C.
- Remove most of the ethanol and air dry on ice for 3−5 min.
NOTE: Ethanol should be removed after this step; however, careful inspection of bottom of the tube is important to ensure no ethanol is left. - After the tube is air dried, add 15−20 µL of RNase-free ddH2O and measure RNA concentration and purity using a spectrophotometer at 260 nm wavelength. The purity of the sample should be measured as a 260 nm/280 nm ratio. This ratio should be approximately >2.
NOTE: Isolated RNA can be frozen and stored at -20 °C. It is, however, highly preferential to use a cDNA synthesis kit as soon as possible to ensure high-quality cDNA production. Quantitative PCR can follow using instructions provided by the manufacturer of the thermocycler used in the laboratory.
Representative Results
The protocol described here allows for specific isolation of unc-17::GFP-positive cholinergic neurons from the roundworm C. elegans for subsequent ex vivo studies such as cell type-specific gene expression profiling and eventual short-term culturing for patch-clamp electrophysiology measurements.
Figure 1 shows unc-17::GFP-positive cholinergic neurons in their normal setting. The unc-17 gene is expressed throughout the C. elegans body and codes for a vesicular acetylcholine transmembrane transporter17. The expression of this gene can be observed in head, midbody, and tail neurons and neuron projections.
Figure 2A shows a pictorial overview of the neuron isolation procedure outlined in the methods section. Step one is to synchronize and culture the worms. Additionally, Figure 2B shows representative fluorescence micrographs of unc-17::GFP-positive neurons after isolation as well as respective negative control from unmodified N2 wild type animals. Isolated cells can stay alive up to 3 days, when supplemented with Leibovitz's L-15 medium and 10% FBS.
Figure 3 shows representative data of qPCR from GFP expressing cholinergic neurons as well as GFP-expressing muscle cells. After successful sorting of cells by either method, RNA was isolated via a phenol and guanidine isothiocyanate method after which cDNA was produced using a synthesis kit. Expression of cholinergic neuron-specific genes, tph-1 and snb-1, a tryptophan hydroxylase and synaptic vesicle transporter, respectively, is elevated in unc-17::GFP isolated cells but is not present in myo-3::GFP isolated cells. The inverse is true with the expression of the muscle-specific myosin heavy chain transcript of myo-2. Expression of the latter gene is limited to isolated muscle cells but not the isolated cholinergic cells.
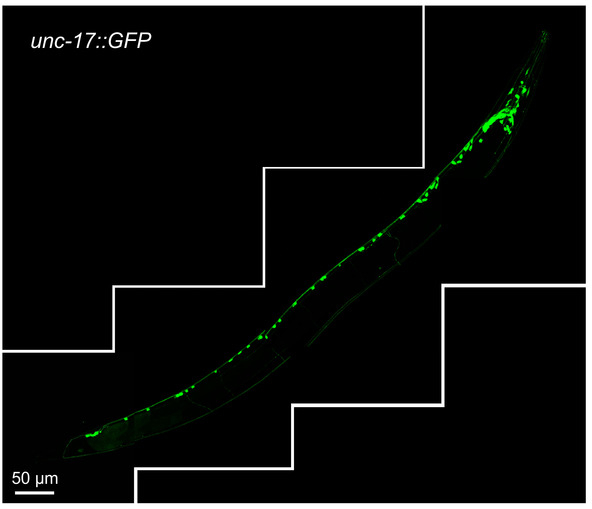
Figure 1: Reconstructed image of cholinergic neurons in one-day-old wild type transgenic animals expressing unc-17::GFP reporter. The image was assembled from four separate confocal fluorescence images of the same worm, covering the length of the entire animal. The scale bar of each individual image is 50 μm. Please click here to view a larger version of this figure.
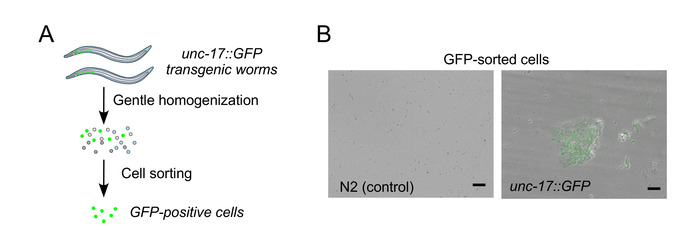
Figure 2: Workflow of cell isolation and representative GFP-expressing cells isolated from unc-17::GFP strain. (A) Schematic workflow depiction for the isolation of specific cell populations enriched in unc-17::GFP-expressing cholinergic neurons from respective C. elegans transgenic strains. (B) Representative fluorescence images showing the presence of GFP signal in the isolated unc-17::GFP-positive cells. Note that the image shows a single focal plane. Scale bar = 10 μm. Figure 2B is reproduced with permission from the Journal of Cell Science and has been published by Germany et al.12. Please click here to view a larger version of this figure.
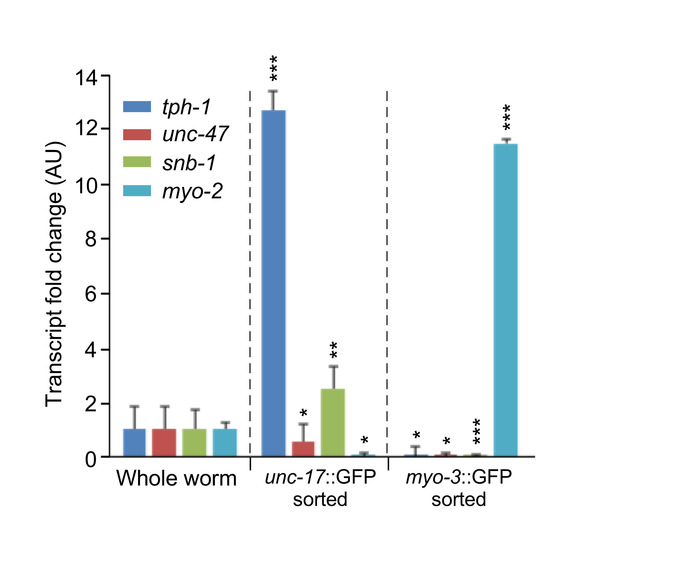
Figure 3: Representative qPCR analysis of unc-17-neuron specific markers (tph-1, unc-47 and snb-1) in whole-animal versus unc-17::GFP reporter cell isolates confirming specific enrichment of cholinergic neurons of interest. Additional negative controls used here were myo-3::GFP reporter-expressing muscle cells, which had been isolated following the same protocol. Data show mean values ± SD (n = 3 biological replicates). *p < 0.05; **p < 0.01; ***p < 0.001 by paired t test. This figure has been modified from Germany et al.12. Please click here to view a larger version of this figure.
Discussion
The roundworm C. elegans is a well-established and powerful model to study neuronal health and disease2. With ample genetic tools to manipulate these animals and manageable quantity of precisely mapped various neuron types, a great deal of data can be collected with a relatively small quantity of material. Here, we outline an optimized method to isolate distinct neurons from whole animals. By disrupting the worm’s outer cuticle proteins, a slurry of various cell types can be isolated, including neurons and muscular cells5. These isolated cells can be used in a multitude of unique experiments. The ability to use either cell sorting through flow cytometry or antibody-labeled magnetic beads allows for an adaptive technique dependent of the parameters of specific experiment. Extracting RNA from a specific C. elegans cell type also allows for a more thorough examination and a mechanistic understanding of various cellular processes.
There are critical steps discussed here which must be followed closely: 1) aging and harvesting well-fed worms from 25 µM FuDR plates; 2) time in SDS-DTT incubation or treatment with protease mixture; 3) selection of GFP-positive cells. First, during aging of the worms, it is imperative to be diligent to ensure that any eggs laid do not hatch, as this will cause age heterogeneity in the collected worms, causing significant variability in data. The wash steps when harvesting worms for isolation are also necessary to ensure that any dead or immature animals are not collected. Second, excessive incubation of worms in the SDS-DTT buffer can cause undesired breakdown of important cellular components and toxicity to the worms, preventing isolation of target cell types. Maintaining cell viability is essential for successful RNA extraction from or further short-term culturing of isolated cells. Because the majority of neuronal cells are delicate, overly harsh treatment during either the SDS-DTT buffer or the protease mixture treatment can result in massive damage to these cells. If a stress response pathway is the target of the experiment, conditions of incubations may be slightly altered as the highly reductive power of DTT may cause enhanced expression of certain stress response genes. In this case, it is important to successfully wash cell pellets with L-15-supplemented medium to halt the reaction and quickly re-supply the cells with nutrients. Finally, carefully choosing which selection step is most appropriate to the cell type of interest will allow for maximum recovery of these cells. Likewise, considering cellular localization of cell type-specific markers may help researchers choose the most optimal cell enrichment approach. Using flow cytometry will yield a more homogenous and viable cell culture, whereas using anti-GFP magnetic beads is less time consuming and labor intensive.
It is important to note that the method described here does have some limitations. First, robust expression of GFP in desired cell types should be verified to ensure suitable isolation by FACS or antibody-coupled magnetic beads. Second, even gentle cuticle disruption can lead to a poor yield or even loss of certain body wall cells, or other low-abundance cell populations such as dat-1 dopaminergic neurons. When choosing cell types, the more abundant cell type can usually provide a more successful yield. Nonetheless, this method can be used for the isolation of less abundant cell type populations as well.
In summary, the procedure discussed in this article presents an effective method of extraction and isolation of individual cell types from the roundworm C. elegans. The method is robust enough to allow for the extraction of RNA, and subsequent gene profiling, and sensitive enough to allow for cultivation of the isolated cells. Although the exact methodology utilized to isolate specific groups of cells may vary between different cell types, the workflow described in this report is generally effective and can be applied to virtually all kinds of worm cells.
Offenlegungen
The authors have nothing to disclose.
Acknowledgements
We thank Dr. Jennifer Fox and the members of the Khalimonchuk laboratory for insightful comments. We acknowledge the support from the National Institutes of Health (R01 GM108975 to O.K. and T32 GM107001-01A1 to E.M.G.).
Materials
| 6-well plate | Fisher Scientific | 12-556-004 | |
| Agar, Molecular Biology Grade | VWR | A0930 | |
| CaCl2 | Sigma | C5670 | |
| Chloroform | Sigma | 496189 | |
| Contess Automated Cell Counter | Invitrogen | Z359629 | |
| DTT | USBiological | D8070 | |
| Ethanol | Decon Labs | 2701 | |
| FBS | Omega Scientific | FB-02 | |
| Fluorodeoxyuridine | Sigma | F0503 | |
| HEPES | Sigma | H3375 | |
| Isopropanol | VWR | BDH1133 | |
| KCl | Amresco | O395 | |
| KH2PO4 | USBiological | P5110 | |
| Kimwipes | Kimberly-Clark Professionals | 7552 | |
| Leibovitz's L-15 Medium | Gibco | 21083027 | |
| MgCl2 | Sigma | M8266 | |
| MgSO4 | USBiological | M2090 | |
| Na2HPO4·7H2O | USBiological | S5199 | |
| NaCl | VWR | X190 | |
| NaOCl (Bleach) | Clorox | ||
| NaOH | Amresco | O583 | |
| Penicillin-Streptomicen | Fisher Scientific | 15140122 | |
| Peptone Y | USBiological | P3306 | |
| Pronase E | Sigma | 7433 | protease mixture from Streptomyces griseus |
| SDS | Amresco | O227 | |
| SMT1-FLQC fluorescence stereomicroscope | Tritech Research | ||
| Sucrose | USBiological | S8010 | |
| SuperScript IV One-Step synthesis kit | ThermoFisher | 12594025 | |
| TRIzol | Invitrogen | 15596026 | phenol and guanidine isothiocyanate solution |
| Trypan Blue Stain | Invitrogen | T10Z82 | |
| α-GFP magnetic beads | MBL | D153-11 |
Referenzen
- Sulston, J. E., Schierenberg, E., White, J. G., Thomson, J. N. The embryonic cell lineage of the nematode Caenorhabditis elegans. Entwicklungsbiologie. 100, 64-119 (1983).
- White, J. G., Southgate, E., Thomson, J. N., Brenner, S. The structure of the nervous system of the nematode Caenorhabditis elegans. Philosophical Transactions of the Royal Society B. 314, 1-340 (1986).
- Zeng, H., Sanes, J. R. Neuronal cell-type classification: challenges, opportunities and the path forward. Nature Reviews in Neuroscience. 18, 530-546 (2017).
- Selkoe, D. J. Cell biology of protein misfolding: The examples of Alzheimer’s and Parkinson’s disease. Nature Cell Biology. 6, 1054-1061 (2004).
- Choi, M. L., Gandhi, S. Crucial role of protein oligomerization in the pathogenesis of Alzheimer’s and Parkinson’s disease. FEBS Journal. 285, 3631-3644 (2018).
- Burman, J. L., et al. Mitochondrial fission facilitates the selective mitophagy of protein aggregates. Journal of Cell Biology. 210, 3231-3247 (2017).
- Shpilka, T., Haynes, C. M. The mitochondrial UPR: mechanisms, physiological functions and implications. Nature Reviews Molecular Cell Biology. 19, 109-120 (2018).
- Haynes, C. M., Petrova, K., Benedetti, C., Yang, Y., Ron, D. ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Developmental Cell. 13, 467-480 (2007).
- Walter, P., Ron, D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 334, 1081-1086 (2011).
- Chen, C., Chen, Y., Jiang, H., Chen, C., Pan, C. Neuron aging: learning from C. elegans. Journal of Molecular Signal. 8 (14), 1-10 (2013).
- Germany, E. M., et al. The AAA-ATPase Afg1 preserves mitochondrial fidelity and cellular health by maintaining mitochondrial matrix proteostasis. Journal of Cell Science. 131, jcs219956 (2018).
- Zhang, S., Banerjee, D., Kuhn, J. R. Isolation and culture of larval cells from C. elegans. PLOS One. 6 (4), e19505 (2011).
- Morley, J. F., Morimoto, R. I. Regulation of longevity in Caenorhabditis elegans by heat shock factor and molecular chaperones. Molecular Biology of the Cell. 15, 657-664 (2004).
- Pereira, L., et al. A cellular regulatory map of the cholinergic nervous system of C. elegans. eLife. 4, e12432 (2015).
