This report describes the full SLIC-CAGE protocol for obtaining sequencing-ready libraries from nanograms of starting total RNA material (Figure 1). To obtain the synthetic RNA carrier mix, first, PCR carrier templates need to be prepared and gel-purified to eliminate PCR side products (Figure 2A). Each PCR template (ten in total) is produced by using a common forward, but a different reverse primer (Table 2), leading to different lengths of the PCR template to enable size variability of synthetic RNA carriers. Once purified, PCR templates are used for in vitro transcription of the carrier molecules. A single RNA carrier product is expected if the templates are gel-purified (see representative gel-analysis in Figure 2B). Preparation of the carrier can be upscaled depending on the need, and when prepared, mixed and frozen at -80 °C for future use.
Using the recommended minimal amount of sample total RNA (10 ng) combined with 16-18 cycles of PCR amplification, high complexity SLIC-CAGE libraries can be achieved. Number of PCR cycles required to amplify the final library highly depends on the amount of total input RNA used (the expected number of cycles is presented in Table 4).
After the first round of degradation, in qPCR results (step 17), the expected difference between Ct values obtained using adaptor_f1 or carrier_f1 primer is 1-2, with Ct values obtained with adaptor_f1 lower than with carrier_f1.
The distribution of the fragment lengths in the final library is between 200-2,000 bp with the average fragment size of 700-900 bp (based on the region analysis using Bioanalyzer software, Figure 4B,D). Shorter fragments, as presented in Figure 4A,C, have to be removed by additional rounds of size-exclusion (steps 20-21). These short fragments are PCR amplification artefacts and not the target library. Note that shorter fragments cluster better on the sequencing flow cells and may cause sequencing problems.
The expected amount of library material obtained per sample is between 5-50 ng. Significantly lower amounts are indicative of sample loss during the protocol. If the obtained low quantity is enough for sequencing (2-3 ng of the pooled libraries is needed), the libraries may be of lower complexity (see below).
Depending on the sequencing machine, quantity of the library loaded onto the flow cell may need to be optimised. Using an Illumina HiSeq 2500, loading 8-12 pM SLIC-CAGE libraries gives on average 150-200 million reads, with >80% of reads passing quality score Q30 as threshold.
The obtained reads are then mapped to the reference genome [for 50 bp reads, Bowtie212 can be used with default parameters that allow zero mismatches per seed sequence (22 bp)]. Expected mapping efficiencies depend on the total RNA input amount and are presented in Table 5. The uniquely mapped reads can then be loaded into R graphical and statistical computing environment13 and processed using CAGEr (Bioconductor package14). The package vignette is easy to follow and explains the workflow and processing of the mapped data in detail. An easy visual control of the library complexity is the distribution of promoter width, as low-complexity libraries will have artificially narrow promoters (Figure 5A, SLIC-CAGE library derived from 1 ng of total RNA, for details see previous publication10). However, even the low-complexity SLIC-CAGE libraries allow identification of true CTSSs, with greater precision than alternative methods for low/medium-input TSS mapping (Figure 5B,C).
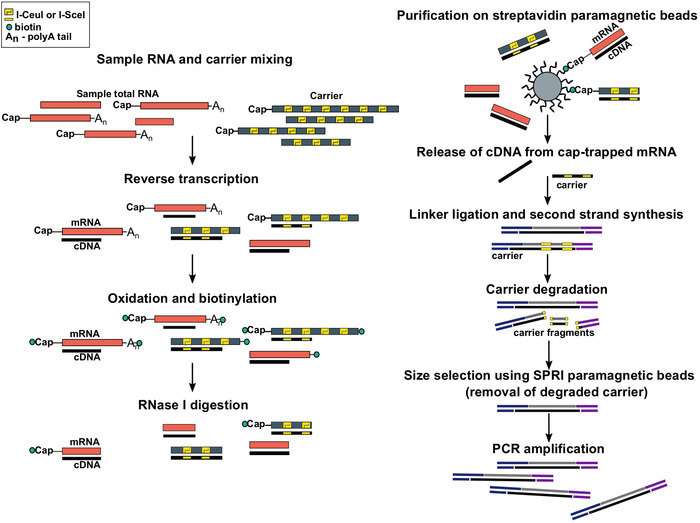
Figure 1: Steps in the SLIC-CAGE protocol. Sample RNA is mixed with the RNA carrier mix to achieve 5 µg of total RNA material. cDNA is synthesised through reverse transcription and the cap is oxidized using sodium periodate. Oxidation allows attachment of biotin to the cap using biotin hydrazide. Biotin gets attached to the mRNA’s 3′ end, as it is also oxidized using sodium periodate. To eliminate biotin from mRNA:cDNA hybrids with incompletely synthesized cDNA and from the 3′ ends of mRNA, the samples are treated with RNase I. cDNA that reached the 5′ end of mRNA is then selected by affinity purification on streptavidin magnetic beads (cap-trapping). After release of cDNA, 5′- and 3′-linkers are ligated. The library molecules that originate from the carrier are degraded using I-SceI and I-CeuI homing endonucleases and the fragments are removed using SPRI magnetic beads. The library is then PCR amplified. Please click here to view a larger version of this figure.
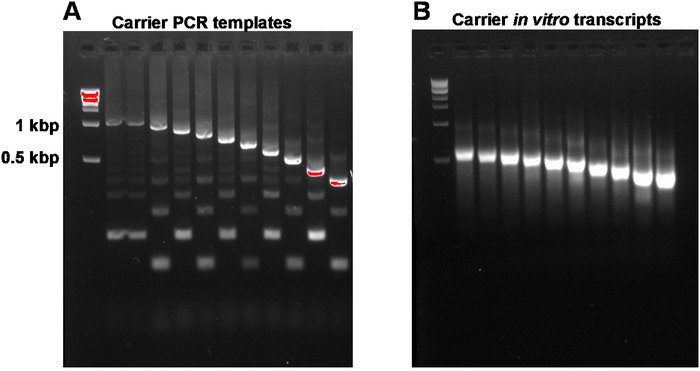
Figure 2: Representative gel-analysis of carrier PCR templates and carrier in vitro transcripts. (A) Carrier PCR templates prior to gel purification: the first well contains the 1 kbp marker, followed by carrier PCR templates 1, 1-10. (B) Carrier in vitro transcripts: the first well contains the 1 kbp marker, followed by carrier transcripts 1-10. Carrier transcripts were denatured by heating for 5 min at 95 °C prior to loading. Please click here to view a larger version of this figure.
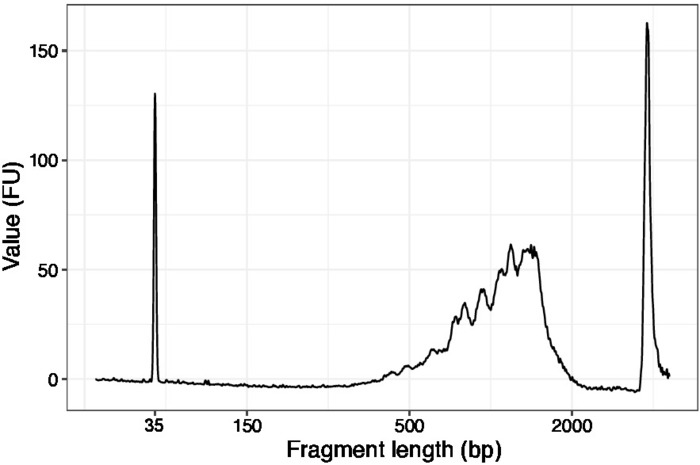
Figure 3: Representative DNA quality (high sensitivity DNA chip) trace of SLIC-CAGE prior to first round of carrier degradation. Please click here to view a larger version of this figure.
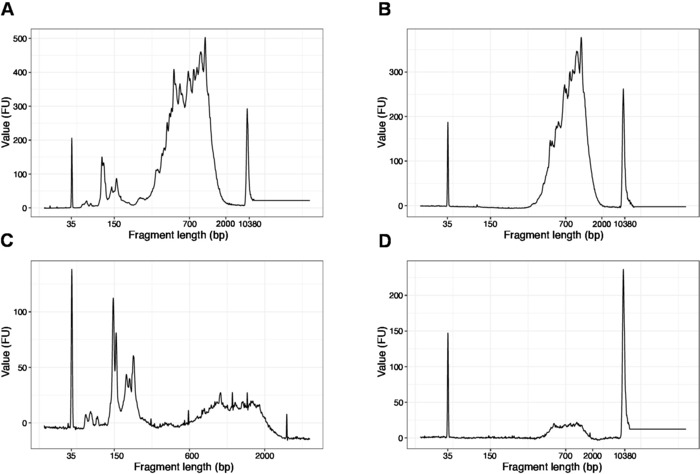
Figure 4: Representative DNA quality (high sensitivity DNA chip) traces of SLIC-CAGE libraries after PCR amplification. (A) SLIC-CAGE library that requires additional size-selection for removal of short fragments. (B) SLIC-CAGE library after size-selection using 0.6x SPRI beads to sample ratio. (C) SLIC-CAGE library of lower output amount that requires size-selection for removal of short fragment. (D) SLIC-CAGE library of lower output amount after size-selection using 0.6:1 SPRI beads to sample ratio. Please click here to view a larger version of this figure.
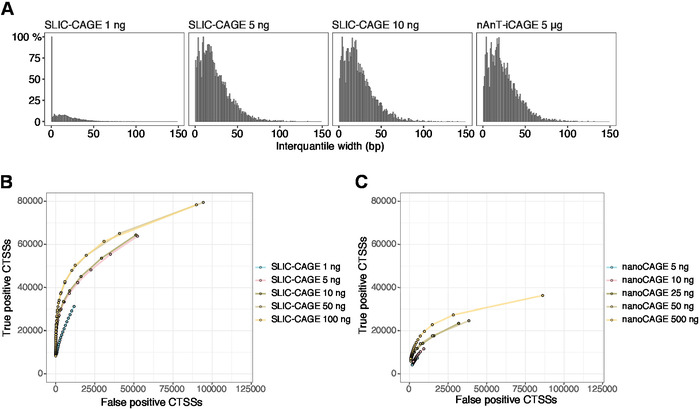
Figure 5: Validation of SLIC-CAGE libraries. (A) Distribution of tag cluster interquantile widths in SLIC-CAGE libraries prepared from 1, 5, or 10 ng of S. cerevisiae total RNA, and in the nAnT-iCAGE library prepared from 5 µg of S. cerevisiae total RNA. A high amount of narrow tag clusters in the 1 ng SLIC-CAGE library indicates its low complexity. (B) ROC curves for CTSS identification in S. cerevisiae SLIC-CAGE libraries. All S. cerevisiae nAnT-iCAGE CTSSs were used as a true set. (C) ROC curves for CTSS identification in S. cerevisiae nanoCAGE libraries. All S. cerevisiae nAnT-iCAGE CTSSs were used as a true set. Comparison of ROC curves shows that SLIC-CAGE strongly outperforms nanoCAGE in CTSS identification. Data from ArrayExpress E-MTAB-6519 was used. Please click here to view a larger version of this figure.
Table 1: Sequence of the carrier synthetic gene. I-SceI sites are bold and italicized in purple, and I-CeuI recognitions sites are green. Please click here to view this table (Right click to download).
| carrier | reverse primer 5’-3’ | PCR product length / bp | |
| 1 | PCR_N6_r1: NNNNNNCTACGTGTCGCAGACGAATT | 1034 | |
| 2 | PCR_N6_r2: NNNNNNTATCCAGATCGTTGAGCTGC | 966 | |
| 3 | PCR_N6_r3: NNNNNNCACTGCGGGATCTCTTTACG | 889 | |
| 4 | PCR_N6_r4: NNNNNNGCCGTCGATAACTTGTTCGT | 821 | |
| 5 | PCR_N6_r5: NNNNNNAGTTGACCGCAGAAGTCTTC | 744 | |
| 6 | PCR_N6_r6: NNNNNNGTGAAGAATTTCTGTTCCCA | 676 | |
| 7 | PCR_N6_r7: NNNNNNCTCGCGGCTCCAGTCATAAC | 599 | |
| 8 | PCR_N6_r8: NNNNNNTATACGCGATGTTGTCGTAC | 531 | |
| 9 | PCR_N6_r9: NNNNNNACCGCCGCGCCTTCCGCAGG | 454 | |
| 10 | PCR_N6_r10: NNNNNNCAGGACGTTTTTGCCCAGCA | 386 | |
| * Forward primer is the same for all carrier templates. Underlined is the T7 promoter sequence. PCR_GN5_f1: TAATACGACTCACTATAGNNNNNCAGCGTTCGCTA | |||
Table 2: Primers for carrier template amplification. Forward primer is the same for all carrier templates. Underlined is the T7 promoter sequence. PCR_GN5_f1: TAATACGACTCACTATAGNNNNNCAGCGTTCGCTA. Using differing reverse primers, PCR templates and hence carrier RNAs of differing length are produced.
| carrier | length | uncapped/µg | capped/µg |
| 1 | 1034 | 3.96 | 0.45 |
| 2 | 966 | 8.36 | 0.95 |
| 3 | 889 | 4.4 | 0.5 |
| 4 | 821 | 6.6 | 0.75 |
| 5 | 744 | 4.4 | 0.5 |
| 6 | 676 | 3.08 | 0.35 |
| 7 | 599 | 4.4 | 0.5 |
| 8 | 531 | 3.96 | 0.45 |
| 9 | 454 | 2.64 | 0.3 |
| 10 | 386 | 2.2 | 0.25 |
Table 3: RNA carrier mix. In total 49 µg of the carrier mix 0.3-1 kbp: uncapped = 44 µg, capped = 5 µg.
| Total RNA input /ng | PCR cycles |
| 1 ng | 18 |
| 2 ng | 17 |
| 5 ng | 16 |
| 10 ng | 15-16 |
| 25 ng | 14-15 |
| 50 ng | 13-15 |
| 100 ng | 12-14 |
Table 4: Expected number of PCR cycles in dependence of sample total RNA input. Approximate number of cycles is based on experiments performed using Saccharomyces cerevisiae, Drosophila melanogaster, and Mus musculus total RNA.
| Total RNA input/ng | % overall mapped | % uniquely mapped | % carrier |
| 1 ng | 30 | 20-30 | 30 |
| 2 ng | 60 | 20-50 | 10 |
| 5 ng | 60-70 | 40-60 | 5-10 |
| 10 ng | 60-70 | 40-60 | 5-10 |
| 25 ng | 65-80 | 40-70 | 0-5 |
| 50 ng | 65-80 | 40-70 | 0-3 |
| 100 ng | 70-85 | 40-70 | 0-2 |
Table 5: Expected mapping efficiency and in dependence of total RNA input amount. Approximate numbers are presented and based on experiments performed using Saccharomyces cerevisiae and Mus musculus total RNA.
Supplementary Table 1: Primer sequences. Please click here to view this table (Right click to download).
Supplementary Table 2: Annealing of 5’ and 3’ linkers. Please click here to view this table (Right click to download).
Supplementary Table 3: 5’ linker annealing. Please click here to view this table (Right click to download).
Supplementary Table 4: 5’ linker mixing. Please click here to view this table (Right click to download).
Supplementary Table 5: 5’ linker dilution. Please click here to view this table (Right click to download).
Supplementary Table 6: 3’ linker annealing. Please click here to view this table (Right click to download).
Supplementary Table 7: 3’ linker dilution. Please click here to view this table (Right click to download).
Supplementary Table 8: Preparation of standard serial dilutions. Please click here to view this table (Right click to download).
