Using the procedure presented here, we tested whether the survival of cortical interneurons during early postnatal stages is regulated by activity in a cell autonomous manner. We performed 3 brain slice electroporation experiments (12-16 embryos [E14.5 embryos] per experiment) with the pCAGGs-IRES-GFP (control) and pCAGGs-hM3D(Gq)-IRES-RFP expression vectors, at a concentration of 1 µg/µL for each construct. In our electroporation experiments, only a fraction (approximately 50%; Figure 3) of the GFP+ neurons co-expressed hM3D(Gq) (RFP+) and therefore the GFP+RFP– population served as an internal control for the effect of DREADD ligands. Transfected cortical embryonic interneurons were mechanically dissociated and the resulting cell suspension (8 x 105 cells/µL) grafted in the cortex of P0-P1 wild type mice. We had performed 6 injections per brain. In each experiment, a minimum of 6 new born pups were injected. Administration of CNO selectively increased the activity of transfected RFP+ cells, as demonstrated by the expression of the activity-dependent protein cFos (Figure 4). CNO treatment according to the described protocol (administer twice daily P14-P17) resulted in an increase in the proportion of GFP+RFP+ relative to GFP+RFP– cells, in comparison to vehicle (0.5% DMSO in saline) administered littermates (Figure 5).
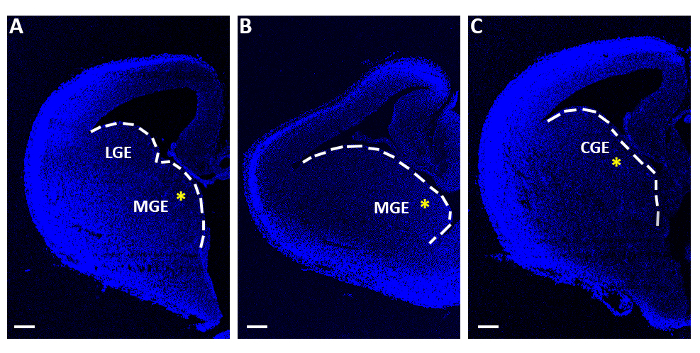
Figure 1: Representative telencephalic slices used for acute electroporation experiments. (A-C) Telencephalic slices obtained at three distinct sequential rostro-caudal levels, stained with 4′,6-diamidino-2-phenylindole (DAPI). LGE: lateral ganglionic eminence; MGE: medial ganglionic eminence; CGE: caudal ganglionic eminence. Scale bars = 200 µm. The yellow asterisks indicate the electroporation site in each slice. The white line marks the edge of the ganglionic eminence. Please click here to view a larger version of this figure.
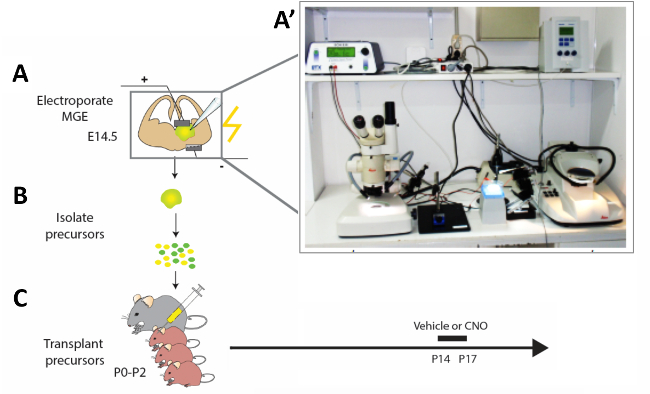
Figure 2: Schematic representation of the experimental workflow. (A) Mouse brain slices are electroporated with appropriate constructs, and (B) after 12 h modified cortical interneuron (CI) precursors are isolated and (C) transplanted in the pallium of newborn mouse pups (P0−P2). In order to modify the activity of immature CIs, P14 pups that had received cell transplantations were injected with CNO or vehicle for four constitutive days according to the presented protocol. (A') Photograph of the acute mouse brain slice electroporation set-up. Please click here to view a larger version of this figure.
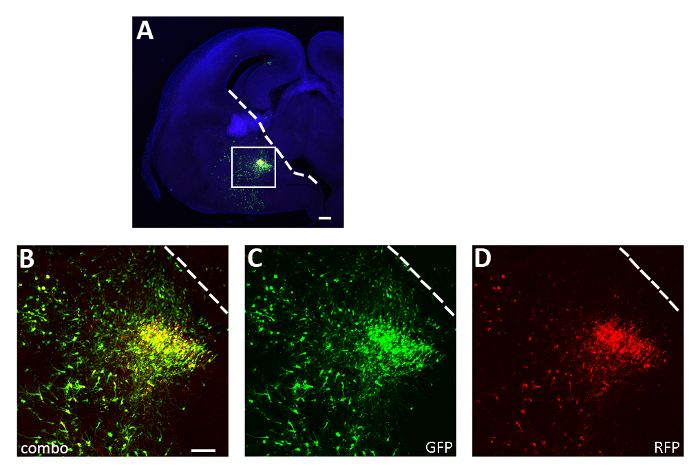
Figure 3: Representative successful acute slice electroporation experiment. (A) Representative coronal section from an E14.5 embryo brain transfected in the CGE with both pCAGGs-IRES-GFP (GFP) and pCAGGs-hM3D(Gq)-IRES-RFP (RFP) plasmids and cultured for 12 h. The section has been immunostained for GFP (A, B, C) and RFP (A, B, D). The boxed area in panel A is magnified to show the expression of both fluorescent reporters (B), GFP (C) and RFP only (D). The white line marks the edge of the ganglionic eminence. B-D: same photo, different channels or combination of the two different channels. Scale bars = 200 µm (A), 100 µm (B-D). This figure has been modified from Denaxa et al.14. Please click here to view a larger version of this figure.
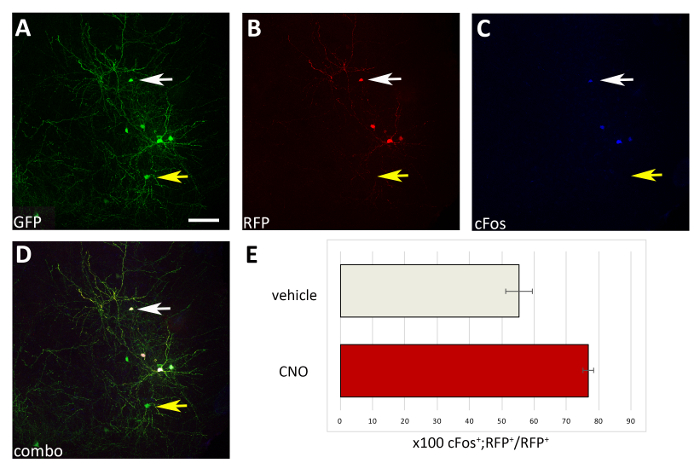
Figure 4: Cell autonomous increase in the activity of M3D(Gq)-expressing transplanted CIs upon CNO administration. (A-D) Representative confocal images of a coronal section of a P17 mouse transplanted at P1 with CI precursors transfected with both pCAGGs-IRES-GFP (GFP) and pCAGGs-hM3D(Gq)-IRES-RFP (RFP) plasmids and treated with CNO. The section has been immunostained for GFP (A), RFP (B), and cFos (C). (D) The combined image of A, B and C immunofluorescence (combo). Note that only CIs co-expressing both plasmids (white arrows in A-D) are also cFos+ compared to CIs expressing only the control-GFP plasmid (yellow arrows in A-D). (E) Quantification of cFos+RFP+ cells found in the pallium of P17 mice transplanted at P1 (normalized to the total RFP+ population) and treated with vehicle or CNO (N = 2). A-D: same photo, different channels, or combination of the three different channels. Scale bars = 50 µm. Please click here to view a larger version of this figure.
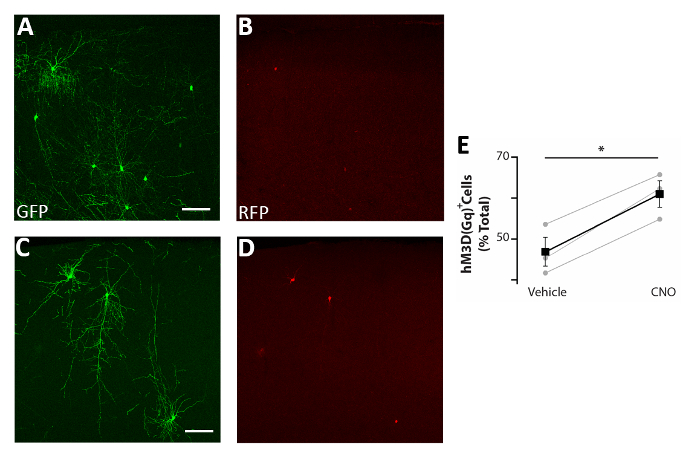
Figure 5: Cell autonomous increase in the activity of CIs enhances survival. (A-D) Representative confocal images of somatosensory cortex coronal slices of P17 mice transplanted at P0-P2 with CI precursors transfected with both pCAGGs-IRES-GFP (GFP) and pCAGGs-hM3D(Gq)-IRES-RFP (RFP) plasmids and treated with vehicle (A-B) or CNO (C-D). (E) Quantification of RFP+ cells found in the forebrain of P17 mice transplanted at P0-P2 (normalized to the total GFP+ population). RFP+(vehicle) = 47% ± 3%, CNO = 61% ± 3%, p = 0.01, Student’s paired sample t test, n = 3 vehicle and 3 CNO, a minimum of 150 cells counted per brain. A and B: same photo, different channels. C and D: same photo, different channels. Scale bars = 50 µm. This figure has been modified from Denaxa et al.14. Please click here to view a larger version of this figure.
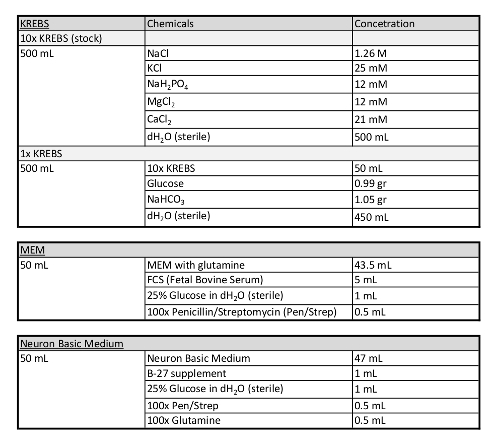
Table 1: Additional information concerning media used in this protocol.
