From 15 mL of fresh whole blood, this protocol generates an average of 1 million CD4+ T cells. These can be frozen for later processing or used immediately. Viability of the CD4+ T cells, fresh or thawed, was consistently >95%. This method of CD4+ T cell isolation allows for flexibility in source material and collection time. This improved ATAC-seq protocol produces a final library of greater than 1 ng/µL for sequencing. Quality control performed using commercially available systems should demonstrate DNA fragments between 200 and 1,000 bp (Figure 2). Sequencing should only be performed with high quality libraries.
All libraries were sequenced to an average depth of greater than 40 million read per sample. While commonly used ATAC-seq protocols have been challenged by contaminating sequencing reads from mitochondrial DNA that can range from 50%-60% of the total sequencing reads1 this improved protocol eliminates the issue. Libraries prepared following this protocol contain on average only 3% mitochondrial reads (Figure 3A). The high percentage of usable reads is sufficiently constant across biological replicates (Figure 3B). The protocol was able to provide highly reproducible results across technical replicates (Figure 4A, B) as well as biological replicates (Figure 4C, D). Additionally, the protocol for CD4+ T cell activation presented takes 48 h rather than one week or more and results in consistent and efficient activation, as demonstrated by reproducible sequencing results (Figure 4A, B). Predicted ATAC-seq peaks are accurately called by the analysis pipeline (Figure 4E). Analysis of sequencing results identified clear changes in chromatin state during human T cell activation. Differentially accessible regions of open chromatin were identified between six samples before and after 48 h of activation (Figure 5).
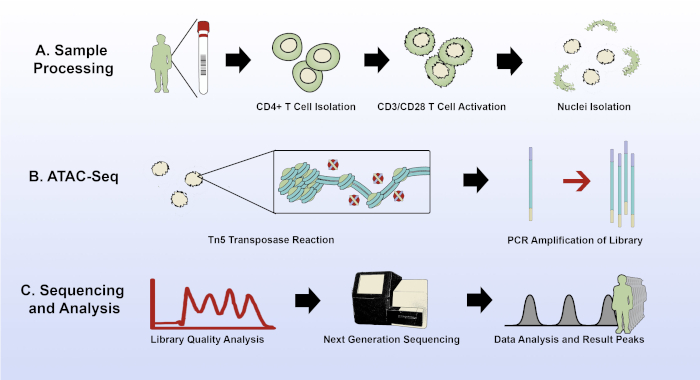
Figure 1: Experimental overview of the modified ATAC-seq protocol. (A) Sample acquisition and processing, from 15 mL of patient whole blood through CD4+ T cell isolation, plating and activation of the T cells, and nuclei isolation with the improved lysis buffer. (B) The transposase reaction and PCR amplification of the sequencing library. (C) Quality analysis, sequencing, and data analysis. Please click here to view a larger version of this figure.
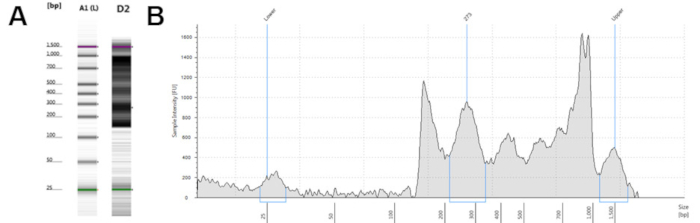
Figure 2: Representative high quality ATAC-seq libraries from a microfluidics-based platform for sizing, quantification and quality control of DNA. (A) Electronic gel image of samples B1 and D1 with banding between 200 and 1,000 base pairs. (B and C) Electropherogram trace result of samples B1 (B) and D1 (C), with peaks between 200 and 1,000 base pairs. Please click here to view a larger version of this figure.
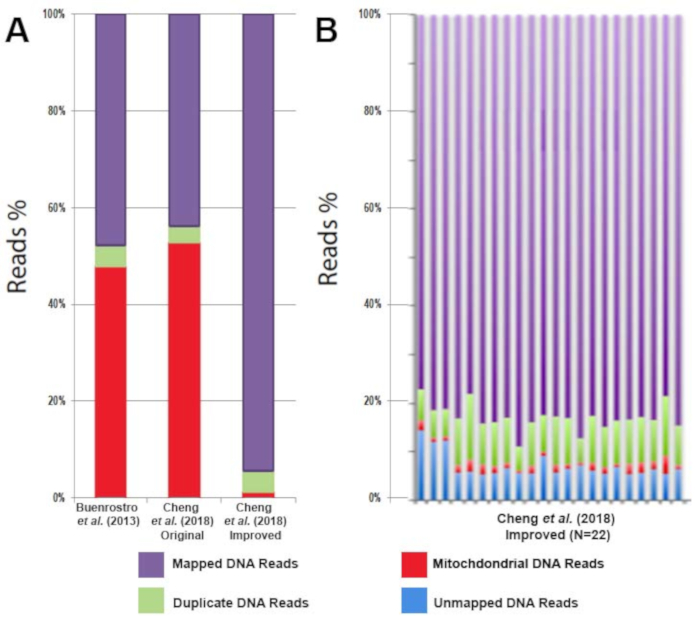
Figure 3: Decreased mitochondrial DNA contamination with the improved ATAC-seq protocol results in an increase in usable DNA sequencing reads. (A) Comparison of usable reads (purple), duplicate reads (green), and mitochondrial reads (red) from CD4+ T cell ATAC-seq profiling in the literature. (B) Comparison of usable reads (purple), duplicate reads (green), mitochondrial reads (red), and unmapped reads (blue) of CD4+ T cell ATAC-seq profiling from multiple healthy individuals (n = 22). This figure has been modified from Cheng et al.10. Please click here to view a larger version of this figure.
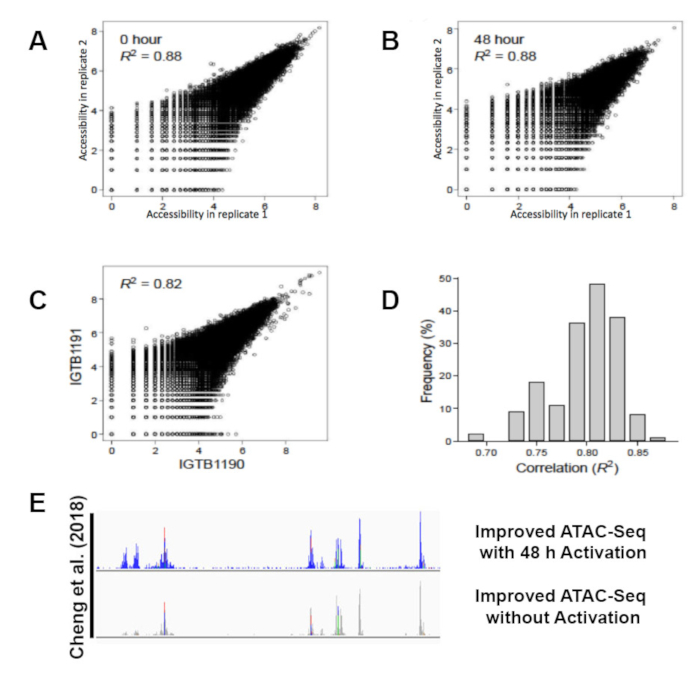
Figure 4: Improved ATAC-seq protocol reproducibility and accuracy. Scatter plots of chromatin accessibility (ATAC-seq signal, x and y-axes) for two replicate experiments of unstimulated (A; 36,486 Th peaks) or activated (B; 52,154 Thstim peaks) T cells demonstrates technical reproducibility. Chromatin accessibility for activated T cells from individuals IGTB1191 (y-axis) and IGTB1190 (x-axis) (C) and histogram of correlations between every pairs of individuals for the 52,154 Thstim peaks (D) demonstrates reproducibility between individuals. (E) ATAC-seq peaks called with our improved ATAC-seq protocol at chromosome 19 Q13.12. This figure has been modified from Cheng et al.10. Please click here to view a larger version of this figure.
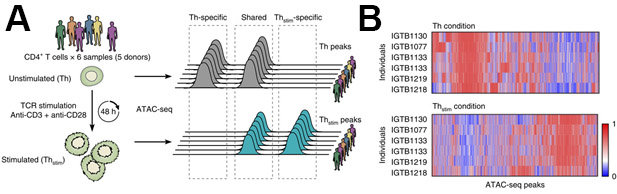
Figure 5: Representative ATAC-seq results of changes in chromatin state after CD4+ T-cell activation. (A) Experimental overview (left) and nomenclature (right). (B) Differentially accessible regions of open chromatin (columns) in six samples (rows) before (top, Th) and after (bottom, Thstim) 48 h activation of primary CD4+ T cells. This figure has been modified from Cheng et al.10. Please click here to view a larger version of this figure.
| 2x TD Buffer | 25 µL |
| TN5 Enzyme | 5 µL |
| Nuclease Free Water | 20 µL |
| Total Volume | 50 µL |
Table 1: Step 3.2 transposase reaction components.
| Ad1_noMX: AATGATACGGCGACCACCGAGATCTACACTCGTCGGCAGCGTCAGATGTG |
| Ad2.1_TAAGGCGA CAAGCAGAAGACGGCATACGAGATTCGCCTTAGTCTCGTGGGCTCGGAGATGT |
| Ad2.2_CGTACTAG CAAGCAGAAGACGGCATACGAGATCTAGTACGGTCTCGTGGGCTCGGAGATGT |
| Ad2.3_AGGCAGAA CAAGCAGAAGACGGCATACGAGATTTCTGCCTGTCTCGTGGGCTCGGAGATGT |
| Ad2.4_TCCTGAGC CAAGCAGAAGACGGCATACGAGATGCTCAGGAGTCTCGTGGGCTCGGAGATGT |
| Ad2.5_GGACTCCT CAAGCAGAAGACGGCATACGAGATAGGAGTCCGTCTCGTGGGCTCGGAGATGT |
| Ad2.6_TAGGCATG CAAGCAGAAGACGGCATACGAGATCATGCCTAGTCTCGTGGGCTCGGAGATGT |
| Ad2.7_CTCTCTAC CAAGCAGAAGACGGCATACGAGATGTAGAGAGGTCTCGTGGGCTCGGAGATGT |
| Ad2.8_CAGAGAGG CAAGCAGAAGACGGCATACGAGATCCTCTCTGGTCTCGTGGGCTCGGAGATGT |
| Ad2.9_GCTACGCT CAAGCAGAAGACGGCATACGAGATAGCGTAGCGTCTCGTGGGCTCGGAGATGT |
| Ad2.10_CGAGGCTG CAAGCAGAAGACGGCATACGAGATCAGCCTCGGTCTCGTGGGCTCGGAGATGT |
| Ad2.11_AAGAGGCA CAAGCAGAAGACGGCATACGAGATTGCCTCTTGTCTCGTGGGCTCGGAGATGT |
| Ad2.12_GTAGAGGA CAAGCAGAAGACGGCATACGAGATTCCTCTACGTCTCGTGGGCTCGGAGATGT |
| Ad2.13_GTCGTGAT CAAGCAGAAGACGGCATACGAGATATCACGACGTCTCGTGGGCTCGGAGATGT |
| Ad2.14_ACCACTGT CAAGCAGAAGACGGCATACGAGATACAGTGGTGTCTCGTGGGCTCGGAGATGT |
| Ad2.15_TGGATCTG CAAGCAGAAGACGGCATACGAGATCAGATCCAGTCTCGTGGGCTCGGAGATGT |
| Ad2.16_CCGTTTGT CAAGCAGAAGACGGCATACGAGATACAAACGGGTCTCGTGGGCTCGGAGATGT |
| Ad2.17_TGCTGGGT CAAGCAGAAGACGGCATACGAGATACCCAGCAGTCTCGTGGGCTCGGAGATGT |
| Ad2.18_GAGGGGTT CAAGCAGAAGACGGCATACGAGATAACCCCTCGTCTCGTGGGCTCGGAGATGT |
| Ad2.19_AGGTTGGG CAAGCAGAAGACGGCATACGAGATCCCAACCTGTCTCGTGGGCTCGGAGATGT |
| Ad2.20_GTGTGGTG CAAGCAGAAGACGGCATACGAGATCACCACACGTCTCGTGGGCTCGGAGATGT |
| Ad2.21_TGGGTTTC CAAGCAGAAGACGGCATACGAGATGAAACCCAGTCTCGTGGGCTCGGAGATGT |
| Ad2.22_TGGTCACA CAAGCAGAAGACGGCATACGAGATTGTGACCAGTCTCGTGGGCTCGGAGATGT |
| Ad2.23_TTGACCCT CAAGCAGAAGACGGCATACGAGATAGGGTCAAGTCTCGTGGGCTCGGAGATGT |
| Ad2.24_CCACTCCT CAAGCAGAAGACGGCATACGAGATAGGAGTGGGTCTCGTGGGCTCGGAGATGT |
Table 2: ATAC-seq oligos designs used for PCR.
| Nuclease Free Water | 11.9 µL |
| 100 µM Custom Nextera Primer 1 (Table 2) | 0.6 µL |
| NEBNext High-Fidelity 2x PCR Master Mix | 25 µL |
| ATAC-Seq Library | 10 µL |
| 25 µM Custom Nextera Primer 2 (Table 2) | 2.5 µL |
| Total Volume | 50 µL |
Table 3: Step 3.5 initial PCR reaction mix.
| CYCLE STEP | TEMPERATURE | TIME | CYCLES |
| Extension | 72 °C | 5 min | 1 |
| Initial Denaturation | 98 °C | 30 s | 1 |
| Denaturation | 98 °C | 10 s | 10 |
| Annealing | 63 °C | 30 s | |
| Extension | 72 °C | 1 min | |
| Hold | 4 °C | Infinity | 1 |
Table 4: Step 3.5 initial PCR amplification cycling program.
| PCR Reaction Aliquot | 5 µL |
| PCR Cocktail from Table 3 with 0.6x Syber Green | 10 µL |
Table 5: Step 3.6 qPCR reaction mix.
| CYCLE STEP | TEMPERATURE | TIME | CYCLES |
| Initial Denaturation | 98 °C | 30 s | 1 |
| Denaturation | 98 °C | 10 s | 20 |
| Annealing | 63 °C | 30 s | |
| Extension | 72 °C | 1 min | |
| Hold | 4 °C | Infinity | 1 |
Table 6: Step 3.6 qPCR cycling program.
| CYCLE STEP | TEMPERATURE | TIME | CYCLES |
| Initial Denaturation | 98 °C | 30 s | 1 |
| Denaturation | 98 °C | 10 s | As Determined |
| Annealing | 63 °C | 30 s | |
| Extension | 72 °C | 1 min | |
| Hold | 4 °C | Infinity | 1 |
Table 7: Step 3.7 PCR cycling program for final PCR amplification.
