This protocol is based on the following resources, which are to be referred for more details and special considerations3,25-28.
1. Density matching of detergent micelles with 2H2O
Note: The density of the buffer solution needs to be matched to the density of the detergent micelles. Common density-adjusting agents include 2H2O, H218O, 2H218O, glycerol and sucrose29. H218O has the same density as 2H2O and may be a better choice if deuteration of exchangeable protons in the protein is not desired. In this procedure, the density of 3-(N,N-dimethylmyristylammonio)-propanesulfonate (C14SB) detergent in 50 mM Tris pH 7.3, 100 mM NaCl will be matched with 2H2O. As an initial guess the following concentrations of 2H2O will be used: 10, 30, and 50% v/v.
1.1. Sample preparation
- Prepare the following stock solutions and filter sterilize through a 0.2 μm syringe filter: 50 mL 500 mM Tris pH 7.3 and 1 M NaCl (10X buffer solution); 1 mL 250 mM C14SB (50X detergent solution).
- Prepare 200 μl of sample solution by mixing 20 μl 10X buffer solution, 4 μl 50X detergent solution, 20 μl 2H2O (99.9%), and 156 μl deionized H2O. Prepare also 200 μl of reference solution by mixing 20 μl 10X buffer solution, 20 μl 2H2O (99.9%), and 160 μl deionized H2O.
- Repeat step 1.1.2 for the other 2H2O concentrations, i.e. 30% and 50%, adjusting the 2H2O and H2O amounts appropriately.
1.2. Assembly of 6-channel AUC cells and sample loading into the cells.
Note: There are two types of AUC cell depending on the sample loading method. Cells without external fill has to be loaded prior to sealing the cell, whereas external-fill cells can be loaded after the cells are sealed. Assembly of an external-fill AUC cell has been described previously3. In this protocol, the assembly of a 6-channel AUC cell without external fill is described. The main difference is that it has screw rings on both sides which need to be tightened separately, and it doesn’t need housing plugs (Fig. 1). The difference in assembly steps are highlighted below.
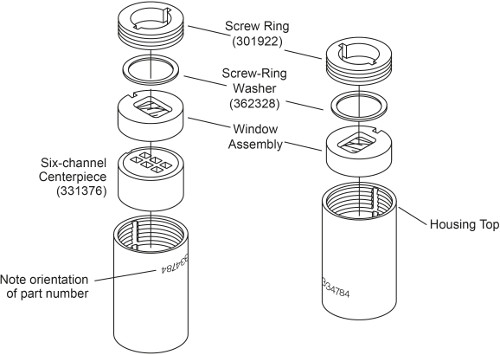
Figure 1. Exploded view of a 6-channel AUC cell without external fill. This figure has been modified from Beckman Coulter An-50 Ti and An-60 Ti Analytical Rotor, Cells, and Counterbalance user manual.
- Prepare two window assemblies for each AUC cell with sapphire window instead of quartz window (Fig. 2). Place the window gasket into the window holder. Slightly bend the window liner and place it into the window holder such that the gap is formed opposite to the window holder keyway. Place the sapphire window inside the window liner, aligning the mark with the window holder keyway.
Note: The quartz window is compressible and thus will produce more light refraction at high speed 28, 30. Therefore for interference measurements above 30000 rpm, such as in this density matching experiment, sapphire windows are used. A sapphire window is heavier than quartz window and has an “X” etched onto its side.
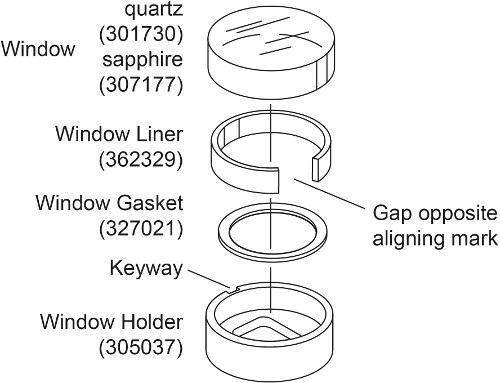
Figure 2. Exploded view of the window assembly. This figure has been modified from Beckman Coulter An-50 Ti and An-60 Ti Analytical Rotor, Cells, and Counterbalance user manual
- Place the cell housing with the part number upside-down. With the keyways aligned with the housing key, slide into the cell housing firstly a 6-sector centerpiece with beveled side down, followed by one window assembly with the window facing down (Fig. 1, left).
- Lightly coat the screw ring threads and screw ring washer with spinkote. Place a screw ring washer on top of the window assembly. Install the screw ring into the window housing with the word “OUT” facing outside. Hand-tighten the screw ring by using the cell aligning tool.
- Using the torque wrench, tighten the screw ring to only 60 inch-pounds.
- Place the cell with the part number upright and positioned at 12-noon. Load 120 μl reference into the left rows and 110 μl sample into the right rows. Ensure that each sample and reference is correctly paired.
Note: Exact sample volume is not critical, but the reference needs to have slightly more volume than the sample (5-10 μl) so that the sample meniscus will be distinct. - Carefully slide into the cell housing one window assembly with the window facing down (Fig. 1, right). Take care not to disturb the cell excessively and spill the contents.
- Repeat step 1.2.3 and 1.2.4, tightening the second screw ring to 120 inch-pounds. Invert the cell and re-tighten the first screw ring to 120 inch-pounds.
- Load the cells into the rotor, install the rotor into the centrifuge and install the monochromator according to the manufacturer’s instructions 28.
Note: Details on this step can also be found in this reference 3.
1.3. Setting up interference measurement
- Start up the user interface software for the AUC instrument and perform laser setup and radial calibration for each cell at 3000 rpm, according to the manufacturer’s instructions, which will be briefly summarized in the following step.
- 1.3.1.1 After sufficient vacuum has been reached (< 100 microns), run the centrifuge at 3000 rpm. Preview the interference pattern in the user interface software and adjust the laser parameters to obtain the highest contrast.
- Create a new set up file (File | New File) specifying “Equilibrium” and “Interference” measurement. Set up a sedimentation equilibrium method (“Method” button) to run at 45000 rpm or the highest speed anticipated for protein samples, whichever is higher, with run temperature at 20°C and collect 1 scan every 15 minutes. Monitor equilibrium progress by opening the data files in HeteroAnalysis and selecting “Match” function after at least 12 hours (approximately overnight, Fig. 3).
Note: a similar function is also available in SEDFIT (Options | Loading Options | Test Approach to Equilibrium).
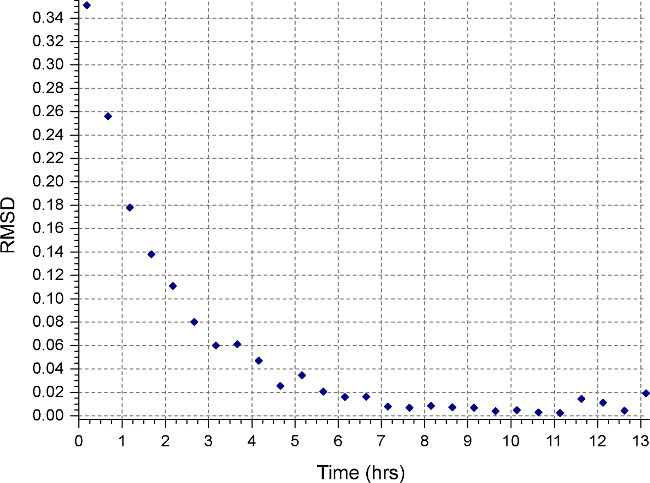
Figure 3. Result from HeteroAnalysis Match function. The Match function can be used to monitor equilibrium progress by comparing RMSD between successive scans and the last scan. This example shows attainment of equilibrium after 8 hr as indicated by RMSD values asymptotic to X-axis.
1.4. Data analysis
- For each set of samples, plot the slope of the radial distribution profile against the 2H2O concentration.
Note: The distribution will be a very shallow exponential that approaches linearity. X-axis intercept corresponds to the matching 2H2O concentration. - For more accurate results, perform the experiment in several replicates. Alternatively, repeat the experiment with a narrower range of 2H2O concentration.
2. Sedimentation equilibrium of SH in C14SB micelles
2.1. Run parameters
- Calculate buffer density and viscosity, protein partial specific volume and centrifugation speed by using SEDNTERP. To calculate buffer density and viscosity, select Compute in “Buffer Data Select” section and enter the buffer components accordingly, including the D2O concentration.
- 2.1.1.1 To calculate protein partial specific volume, select Compute in “V-bar” section and enter the protein amino acid sequence. Specify the highest expected oligomeric size in “Make an oligomer from this monomer: N=” field, in this case N = 5. Calculate the speed by entering values in the RPM field on the main window until σ ≈ 1; this is a rule of thumb to ensure a good exponential shape of the radial distribution profile25.
Note: The values calculated for this experiment was as follows: ρ = 1.03839 g/ml, η = 1.0267 cP = 0.7569 ml/g, ω1 = 16000 rpm. - Calculate subsequent speeds to follow to ensure sufficient difference between the distribution profile at one speed and the next25.
Note: this can also be done from the function "Estimate equilibrium rotor speeds function" in SEDFIT, which takes into account the solution column (filling volume).
2.2. Sample preparations
- Prepare 1 ml reference solution with 5 mM C14SB and 32.3% 2H2O as determined from density matching experiment (section 1), by mixing 100 μl 10X buffer solution (step 1.1.1), 20 μl 50X detergent solution (step 1.1.1), 323 μl 2H2O (99.9%) and 527 μl deionized H2O.
- Dissolve lyophilized, HPLC-purified SH peptides (expression and purification described previously31) in appropriate solvent such as methanol or 50% v/v aqueous acetonitrile. Measure A280 of the dissolved peptides in a microlitre-scale UV/Vis spectrophotometer and aliquot for three samples to give A280, 12mm = 0.3, 0.5, and 0.8 (A280, 10mm = 0.25, 0.417, and 0.67) each when diluted to 130 μl. Lyophilize the samples overnight and resuspend in 130 μl reference solution (step 2.2.1) to give the sample solutions.
Note: SH protein can be detected from UV/Vis absorbance at 280 nm because it contains Trp and Tyr residues. Proteins without aromatic residues can be detected by tagging them with a suitable chromophore, using a Trp-containing mutant, or by using interference measurements instead of absorbance. - Follow steps in section 1.2 to assemble a 6-channel AUC cell with quartz windows. Load the highest concentration sample (A280, 12mm = 0.8) in the channel nearest to the rotor center and lowest concentration sample (A280, 12mm = 0.3) furthest from the rotor center.
2.3. Setting up absorbance measurements
- Create a new set up file (File | New File) specifying “Equilibrium” and “Absorbance” measurement. Specify 280 nm as the detector wavelength.
- Perform radial calibration at 3000 rpm by checking “Radial calibration before first scan” in Scan Options, specifying data collection at low resolution, e.g. with Radial step size = 0.01 cm, Replicates = 3 (low resolution, fast), and executing a Single Scan. After the scan is completed, uncheck the option.
- Set up a sedimentation equilibrium method (“Method” button) to run at the first speed calculated in step 2.1.3, at 20°C, and collect 1 scan every 30 minutes. In each cells’ “Detail”, specify data collection at low resolution as in step 2.3.2. Monitor equilibrium progress by opening the data files in HeteroAnalysis and selecting “Match” function after at least 18 hours (approximately overnight, Fig. 4).
Note: attainment of equilibrium may take substantially longer time for the first speed, whereas subsequent speeds will take less time.
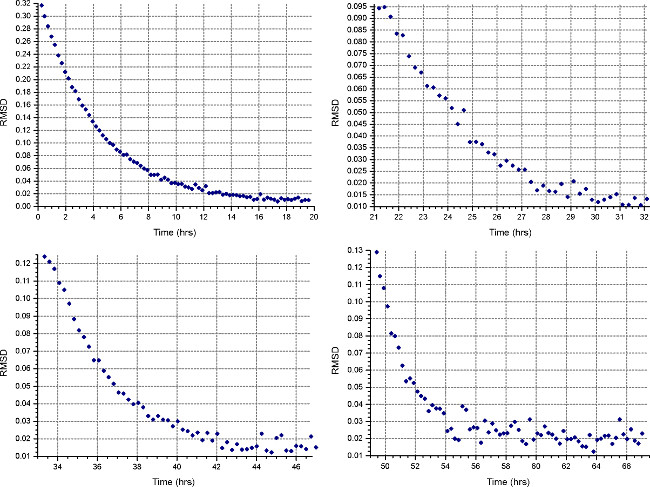
Figure 4. Results from HeteroAnalysis Match function. The first and second speeds (top left and right) appear to have reached equilibrium, but it is better to wait a few more hours to be sure. In comparison, the third and fourth speeds (bottom left and right) have clearly reached equilibrium in a shorter time. Please click here to view a larger version of this figure.
- Once equilibrium has been reached, collect a single scan at high resolution, e.g. with Radial step size = 0.001 cm, Replicates = 10 (high resolution, slow).
- After the scan is completed, repeat step 2.3.3 and 2.3.4 for the next speed.
- Optionally, when the time required to reach equilibrium for each speed is known (calculated or from experience), set up the sedimentation equilibrium method scan to include all the speeds calculated in step 2.1.3 and to collect 1 scan after the equilibration time for each speed. In this case, specify high resolution data collection in each cells’ “Detail”.
2.4. Data analysis in SEDFIT and SEDPHAT
Note: For further details and considerations in data analysis reader is referred to the following website: www.analyticalultracentrifugation.com.
- Open high resolution scans in SEDFIT (Data | load sedimentation equilibrium data) and split the data into 3 channels (these correspond to different concentrations for each sample; Options | Loading Options |Save 6-channel Raw Data in 3 Subsets).
- Re-open data files that belong to the same sample and same concentration but different speeds in SEDFIT. Adjust the meniscus (vertical red line), cell bottom (vertical blue line) and fitting limits (vertical green lines), and export the data for use in SEDPHAT (Data | Export Data to SEDPHAT). Input the parameters calculated in step 2.1.1, as well as rotor type and centerpiece type as requested. Repeat this step for every sample and concentration.
- Open all data from the same sample (all concentrations and speeds) in SEDPHAT and fill in Experiment Parameters; an example is shown in Fig. 5.
Note: when D2O is added in the buffer, deuteration of exchangeable protons could significantly alter the molecular weight of the protein, especially for water-soluble proteins. Membrane proteins, especially small ones like SH protein, are less affected because membrane-embedded regions are protected from exchange. To correct this, input the “buffer D mol fraction”.
Note: At this step it is recommended to save the edited dataset separately by selecting menu Data | Copy All Data And Save As New Config. - Select a Model and fill in Global Parameters for that model.
Note: As an example, the “Monomer-n-Mer Self-Association” model and its parameters are shown in Fig. 6.
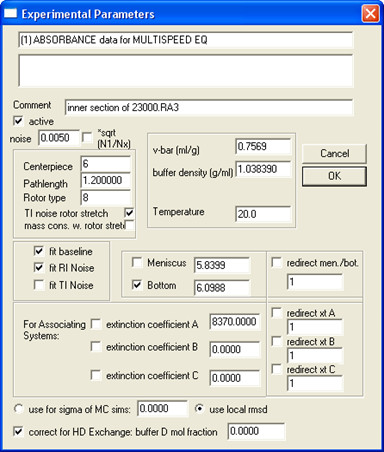
Figure 5. An example on how to fill in Experimental Parameters.
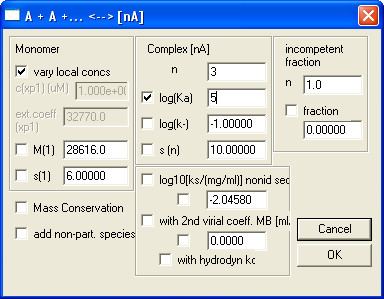
Figure 6. An example on how to fill in Global Parameters for Monomer-n-Mer Self-Association model.
- Run a Global Fit by selecting menu Fit | Global Fit and wait until the fit converges. Note down (or take a screenshot of) the fitting results, especially the global reduced chi-square and log Ka values. Extract other data such as fit data and fitting residuals from menu Copy and Display | Display Thermodynamic Information.
- Return to Global Parameters and check M(1) to fit the monomer molecular weight and repeat step 2.4.5. Note down the fitted molecular weight and global reduced chi-square.
- Repeat step 2.4.3 to 2.4.6 for each model to be tested and compare the fit quality of each model by comparing the reduced global chi-square value as well as fitting residuals.
Note: Small and random fitting residuals generally indicates a good fit, and the model that fits best would have the smallest global reduced chi-square. The fitted monomer molecular weight and its chi-square value should not differ substantially from that of the fixed (theoretical) molecular weight. - Calculate the confidence interval for the obtained log Ka by value by first selecting Statistics | Critical chi-square for error surface projections and inputting the desired confidence interval. Next, go to Statistics | Generate 1-dimensional error surface projection and unselect log Ka in Global Parameters dialog to obtain the chi-square values for log Ka.
Note: readers are advised to consult the following sources (http://www.analyticalultracentrifugation.com/ sedphat/statistics.htm) for more details on the method 32 as well as illustration of this method 33.
The radial distribution profile of C14SB detergent micelles in 50 mM Tris, 100 mM NaCl pH 7.3 forms a very shallow exponential that could be fitted to a linear model (Figure 7A). The slope of this distribution is inversely correlated to D2O concentration (Figure 7B). The point where the slope is zero, i.e., the matching D2O concentration, was found to be 32.3%.
See Figure 7 Below.
The same experiment was repeated for different buffers: 50 mM sodium phosphate, 100 mM NaCl pH 5.5 and 50 mM phosphate-citrate, 100 mM NaCl pH 3 to obtain matching D2O concentrations of 30.3% and 41.0%, respectively.
Samples of SH wild type (wt) in detergent were exposed to pH 3, 5.5 and 7.3 (total 6 samples), followed by centrifugation at 15,000, 19,000, 23,000, 28,000, 34,000 and 42,000 rpm. Data obtained at lower speed could not be reliably fitted, possibly because equilibrium had not been attained, therefore data obtained from the four higher speeds were used. At pH 7.3, SH WT was found to form pentamers (Figure 8 and Table 1) with apparent log Ka = 21.35 (Table 2). The association constant did not change at pH 5.5, but it was significantly reduced at pH 3. This is consistent with previous reports24, which reported a decrease of conductance at lower pH, with a pK of 4.5.
See Figure 8 Below.
See Table 1 Below.
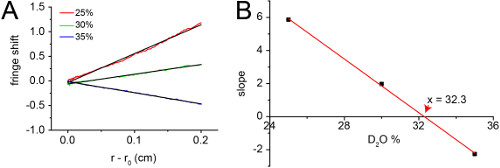
Figure 7. Density matching of C14SB with D2O. (A) Radial distribution profile formed by C14SB detergent micelles in 50 mM Tris, 100 mM NaCl pH 7.3 with 25, 30, and 35% D2O. The data was separately fitted to a linear model (black). (B) The slopes were plotted against D2O concentration (black squares) and fitted to a linear model (red line). The matching D2O concentration is indicated (red arrow).
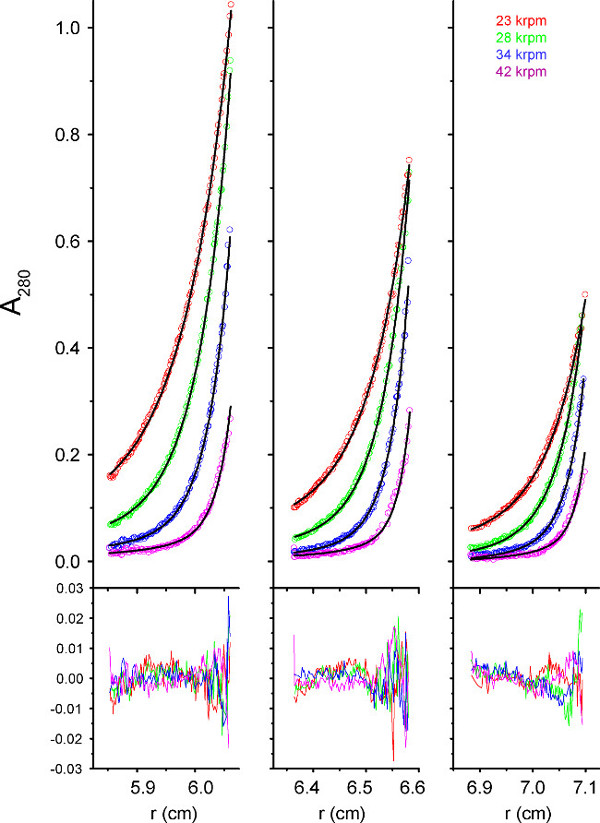
Figure 8. Fitting of SH in C14SB to monomer-pentamer model. Radial distribution profile of SH in C14SB at pH 7 (open circles) was found to fit best to the monomer-pentamer self-association model (black solid line). Fitting residual is shown below. Please click here to view a larger version of this figure.
| Model (n-mer) | Chi-square (fixed MW) | Chi-square (fitted MW) | Fitted MW |
| 3 | 15.444 | 1.0492 | 13477 Da |
| 4 | 3.8094 | 1.0469 | 9889 Da |
| 5 | 1.0499 | 1.0497 | 7822 Da |
| 6 | 2.5994 | 1.547 | 6504 Da |
| 7 | 6.1667 | 1.2112 | 5743 Da |
Table 1. Comparison of global reduced chi-square values of different monomer-n-mer models.
| pH | Lower limit (1σ) | Log Ka | Upper limit (1σ) |
| 3 | 17.432 | 17.576 | 17.737 |
| 5.5 | 20.064 | 20.419 | 20.839 |
| 7.3 | 20.687 | 21.052 | 21.492 |
Table 2. Comparison of apparent log Ka values at different pH.
