Production and Targeting of Monovalent Quantum Dots
Summary
We provide detailed instructions for the preparation of monovalent targeted quantum dots (mQDs) from phosphorothioate DNA of defined length. DNA wrapping occurs in high yield, and therefore, products do not require purification. We demonstrate the use of the SNAP tag to target mQDs to cell-surface receptors for live-cell imaging applications.
Abstract
The multivalent nature of commercial quantum dots (QDs) and the difficulties associated with producing monovalent dots have limited their applications in biology, where clustering and the spatial organization of biomolecules is often the object of study. We describe here a protocol to produce monovalent quantum dots (mQDs) that can be accomplished in most biological research laboratories via a simple mixing of CdSe/ZnS core/shell QDs with phosphorothioate DNA (ptDNA) of defined length. After a single ptDNA strand has wrapped the QD, additional strands are excluded from the surface. Production of mQDs in this manner can be accomplished at small and large scale, with commercial reagents, and in minimal steps. These mQDs can be specifically directed to biological targets by hybridization to a complementary single stranded targeting DNA. We demonstrate the use of these mQDs as imaging probes by labeling SNAP-tagged Notch receptors on live mammalian cells, targeted by mQDs bearing a benzylguanine moiety.
Introduction
The dynamics of single molecules on live cells contributes to their biological function. Single molecule fluorescence imaging is a popular method to study single molecule dynamics on the cell surface1,2,3. However, the most commonly used imaging probes in these studies have several important disadvantages. For example, conventional organic dyes and fluorescent proteins provide moderate brightness, about 105–106 M-1 cm-1, but are photochemically unstable, bleaching after the emission of about 105–106 photons under typical live-cell imaging conditions4,5. In contrast, semiconductor nanoparticles, frequently called quantum dots (QDs), are significantly brighter and more stable, with extinction coefficients in the range of 106–107 M-1 cm-1 and exceeding 107–108 emitted photons before photobleaching5. The improved brightness and photostability of QDs over organic fluorophores enables the observation of single molecules at significantly faster frame rates and over much longer trajectories6.
Despite their advantages and commercial availability, several liabilities remain for these powerful imaging agents. First, they have poorly defined targeting valency, which may result in crosslinking of targeted biomolecules6. Second, they generally have a large hydrodynamic size (> 20 nm) that limits accessibility to certain crowded cellular environments7. Third, they have limited targeting modularity7. Several strategies have attempted to address these problems8,9,10, but generally require specialized knowledge and reagents to implement.
To address these problems, we recently reported a “Steric Exclusion” strategy for preparing monovalent, small, and modular QDs11. The QDs are wrapped with a single long phosphorothioate DNA (ptDNA) polymer. The ptDNA binds to the QD surface through multiple Zn-S interactions between surface-exposed Zn atoms and the phosphorothioate groups of the ptDNA polymer. A single bound polymer sterically and electrostatically excludes the binding of additional equivalents of the polymer without significantly increasing the particle’s overall size (about 2 nm). All reagents are commercially available, products are formed in high yield, and the process requires only desalting steps for purification. Once labeled, QDs wrapped with a single ptDNA (mQDs) bind to complementary DNA strands bearing targeting domains (e.g., benzylguanine (BG), benzylcytosine, or alkylhalides).
These functionalities target the mQDs specifically to enzymatic tags such as SNAP, CLIP & HALO that are genetically fused to the protein of interest. This is a protocol for the synthesis, targeting, and live-cell imaging of mQDs produced by steric exclusion.
Protocol
1. Production of Monovalent Quantum Dots
- Phase transfer of QDs from organic to aqueous phase
- Dilute 200 µl of a 1 µM solution of organic phase QDs with 400 µl of chloroform in a 5 ml glass vial.
- Mix 400 µl of a 0.3 M tetrabutylammonium bromide (TBAB) chloroform solution with 36 µl of neat mPEG thiol (CH3O(CH2CH2O)6C2H5SH) and shake O/N.
- Add 800 µl of a 0.2 M NaOH aqueous solution and shake for 30 sec. A phase transfer occurs within a few minutes, indicated by the transfer of the colored particles to the aqueous phase above the denser organic phase.
- If the particles aggregate in a third phase between the aqueous and organic phases, increase the incubation time with the mPEG thiol. If the aqueous phase remains clear, the QDs did not phase transfer (see Figure 3A). Alternately, increase the concentration of mPEG thiol in step 2 to alleviate poor phase transfer.
- Recover the (colored) aqueous phase and then concentrate the collected QDs with a Centricon spin column (30 kDa molecular weight cut-off) to 1 ml.
- Add the concentrated QD solution into a Sephadex NAP10 column pre-equilibrated with 10 mM Tris buffer containing 30 mM NaCl (pH 8.0). Elute the QDs with 1.5 ml of elution buffer by gravity flow.
- Measure the concentration of QDs with absorption spectroscopy at 350 nm.
- Preparation of mQDs
- Purchase (or synthesize) ptDNA. This protocol uses the sequence 5’-AS50(CT)10(ACTG)5 -3’ (see Table 1).
| DNA | Sequence |
| 5’-AS50(CT)10(ACTG)5-3' | ASASASASASASASASASASASASASASASAS ASASASASASASASAS ASASASASASASASASAS ASASASASASASASASASASASASASASASASAS CTCTCTCTCTCTCTCTCTCTACTGACTGACTGACTGACTG |
| 5’-NH2-(CT)10(CAGT)5-3' | NH2-/C6spacer/ CTCTCTCTCTCTCTCTCTCTCAGTCAGTCAGTCAGTCAGT |
| BG-DNA | BG-/C6spacer/ CTCTCTCTCTCTCTCTCTCTCAGTCAGTCAGTCAGTCAGT |
Table 1. DNA sequences used to produce and target mQDs.
- Prepare 1 ml of 100 nM QD solution in 10 mM Tris buffer containing 30 mM NaCl (pH 8).
- Add drop wise 500 µl of the 100 nM ptDNA solution to the aqueous QDs for 1 min while vigorously stirring. Stir or place on a shaker for an additional 9 hr.
Note: This should theoretically produce a 1:2 ratio of ptDNA:QD. However, the actual stoichiometry is never perfect — usually the result is closer to 1:1.5. In order to produce a 1:1 ratio of ptDNA:QD, densitometry after gel electrophoresis is necessary to quantify the exact ratio of conjugation given the known volumes used above. - Remove ~10 µl of the QD mixture to run on an analytical agarose gel. Also remove a similar concentration of unconjugated QDs in aqueous phase (from step 1.2.1).
- Add ~2 µl of 6x Ficoll loading buffer (to increase solution density) and run these two samples together on a 0.8% wt/v agarose gel in sodium borate buffer for 15 min at 150 V. A single band in the unconjugated control lane migrating close to the well, and two bands in the lane with conjugated QDs are seen (see gels in Figure 2B).
- Calculate the unconjugated QD fraction using the relative intensity of the two bands (see the equation in Figure 2B). Then use that fraction to calculate the additional volume of 100 nM ptDNA solution required to match the number of QDs.
- Repeat steps 1.2.3 to 1.2.5 once more with this calculated volume or until the conjugated QDs collapse into a single band on the gel indicating complete conjugation of all QDs.
- Add 100 µl of 10 mM (CO2H)CH2O(CH2CH2O)6C11H23SH (carboxy PEG6 alkane thiol) in 10 mM Tris buffer containing 30 mM NaCl (pH 8) to the conjugated mQDs produced above, and shake for 10 min.
Note: These PEG ligands passivate the QD surface. A longer PEG will better-passivate the QDs, however, it will also increase their size. The hydrophobic alkane thiol functionality has a much higher affinity for the QDs, in aggregate, than the less hydrophobic PEG-thiol used for phase transfer. Therefore, do not allow this step to proceed too long or the ptDNA will be replaced by the alkane-PEG-thiol. After a ~30 min incubation, a significant fraction of DNA will have been displaced from the QDs (Figure 3B). - To remove excess alkane PEG-thiol, add 0.5 ml of the QD solution to a sephadex NAP5 column pre-equilibrated with elution buffer (10 mM Tris buffer containing 30 mM NaCl). Collect the mQDs with 1.0 ml of elution buffer by gravity flow.
- Concentrate the collected QDs with a Centricon spin column (30 kDa). Store these mQDs at 4 °C for months. Do not freeze them.
Note: If a colored pellet in the bottom of the tube is seen, the dots have aggregated and are likely no longer useable.
2. Production of Targeting (Benzylguanine-) DNA
- Produce or purchase amine-terminated DNA. This protocol uses the sequence 5’-NH2-(CT)10-(CAGT)5-3’ (see Table 1). The poly-CT linker increases accessibility to functionality buried deep in the glycocalyx but may not be needed for all applications. If there is access to a DNA synthesizer, produce amino-modified DNA using an appropriate 5’ amino-modifier (5’ amino-modifier C6).
- Dissolve BG-N-hydroxysuccinimide (BG-NHS) in dry dimethyl sulfoxide (DMSO) at a concentration of 10 mg/ml.
Note: BG-NHS will absorb water from the air and lead to hydrolysis of the reactive NHS-ester, particularly in hydroscopic solvents like DMSO and dimethylformamide (DMF). BG-NHS is best stored at -80 °C as a powder in its own vial within a secondary container containing desiccant. Warm to RT before opening the vial. Alternately, immediately aliquot for single use the BG-NHS ester into a dry polar solvent and freeze at -80 °C, or react fully with the amine DNA in step 2.2. - React by mixing the BG-NHS in DMSO from step 2.1 with amine-terminated DNA in HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) buffered (pH 8.5) water. In a typical react 20 µl of 10 mg/ml BG-NHS in DMSO with 2 µl of 2 mM NH2-(CT)10-(CAGT)5 in 58 µl deionized water buffered with 20 µl 0.5 M HEPES pH 8.5. Carry out reactions in a 1.5 ml microcentrifuge tube. Mix quickly as the reaction proceeds rapidly and must compete with NHS-ester hydrolysis.
- Sonicate for 5–10 min to ensure thorough mixing. Incubate ~1 hr at RT. The stoichiometric ratio of BG:DNA can be altered to drive the reaction to completion.
- Desalt the reaction using a standard NAP5 column into 100 mM triethylamine acetate (TEAA, pH 7).
- Purify the BG-DNA from the unreacted amine-DNA by reversed-phase HPLC running a 100 mM TEAA:acetonitrile (ACN) gradient between 8% and 95% acetonitrile over 30 min. The unreacted amine-DNA will elute before the BG-DNA. Collect the BG-DNA fraction in a conical tube.
- Use a C18 Zorbax column for standard oligonucleotide purification and the following gradient:
0→15 min: 8→30% ACN;
15→21 min: 30–90% ACN;
21→25 min: 90→8% ACN;
25→30 min: 8% ACN;
- Use a C18 Zorbax column for standard oligonucleotide purification and the following gradient:
- Freeze in liquid nitrogen and lyophilize the collected fraction. Resuspend the DNA in deionized water. To be extra careful, lyophilize two more times to remove residual TEAA. Residual TEAA can be toxic to cells at higher concentrations.
- Resuspend the lyophilized BG-DNA in water, making sure to wash the sides of the tube, and dilute to ~25 µM stock. Aliquot and store at 4 °C. Samples were stored for ~9 months without noticeable degradation.
3. Labeling Live Cells with Monovalent Quantum Dots
- Optionally, passivate the mQDs just prior to an imaging experiment using reagents such as casein (0.5%) or bovine serum albumin (BSA, 1–3%).
- Plate cells expressing the SNAP-tagged protein on total internal reflection fluorescence (TIRF)-quality glass. In this experiment, we use a U2OS cell line expressing a SNAP-tagged Notch1 receptor and high quality glass surfaces.
- After the cells have attached to the glass (~24 hr), remove growth media, wash with PBS, and incubate the cells for 10–30 min at RT in ~100–150 µl of PBS or cell media containing ~1 µM BG-DNA from step 2.6 above.
- After incubation with BG, carefully wash the cells with PBS or cell media. Incubate the cells for ~5–10 min with the mQDs produced in step 1.2.9 (and optionally passivated in step 3.1).
- Optionally, wash the unbound mQDs and return the cells to a buffer/media suitable for both imaging and culture. For cells that were to be imaged in TIRF mode, a final wash step was often unnecessary as unbound mQDs in solution diffused so rapidly compared with bound mQDs as to be undetectable during analysis.
4. Microscopy & Analysis
- Image the cells using a TIRF microscope.
Note: The scope must have separable excitation and emission filters, or a custom QD filter cube. QDs are best excited with a low-wavelength laser (405 or 488 nm). QDs of different diameter have characteristic emission wavelengths, typically associated with a large Stoke’s shift. - Because of the brightness of mQDs, collect images at high frame rates in TIRF while imaging the basal side of a live cell.
Note: Alternatively, single-molecule dynamics can be followed at other focal planes using a spinning disk confocal microscope. Automated single-particle tracking software is generally successful at accurately tracking the bright and photostable mQDs.
Representative Results
The phase transfer of the QDs from an organic to an aqueous phase is critical for the production of mQDs, but can be both condition- and QD-specific. Phase transfer in section 1.1 should appear as clean as the first two vials in Figure 3. If transfer appears more like vial 3, then one should try again with different conditions.
Once the QDs are coated with the negatively-charged DNA they should migrate on a gel separately from the non-wrapped QDs. Using an aliquot of unwrapped QDs as a control, a second, faster-migrating band should appear upon addition of the ptDNA, as seen in the first gel in Figure 2B. Complete formation of mQDs is demonstrated with the loss of the immobile band, and its collapse into the mobile band as seen in the last gel in Figure 2B. If an aliquot of your mQD product migrates as a single band separable from the unwrapped QDs, then your mQDs are monovalent and ready to be used in further steps.
Cell labeling should be specific to the target of your choice and should be dependent on protein expression levels and mQD concentration. Empirical optimization of both mQD concentration and mQD passivation is often required for any given application. Figure 4B demonstrates the labeling of U2OS cells expressing a SNAP-tagged Notch receptor with mQDs ranging in concentration from 0.5 nM to 10 nM.
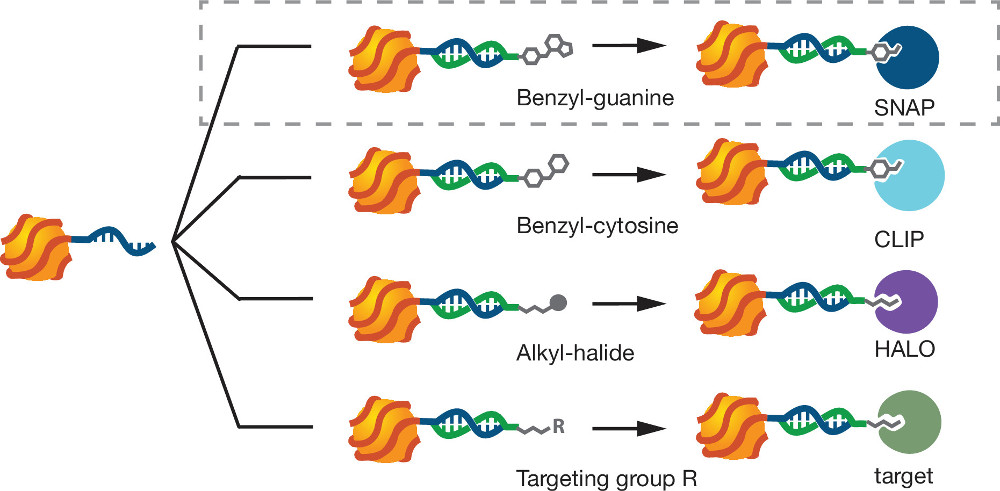
Figure 1. Modular targeting of mQDs. mQDs hybridize to ssDNA functionalized by various targeting molecules. Benzylguanine-linked DNA targets mQDs to SNAP-tagged fusion proteins. Other 5’ modifications and DNA sequences enable the targeting of mQDs to a variety of other biomolecules. Please click here to view a larger version of this figure.
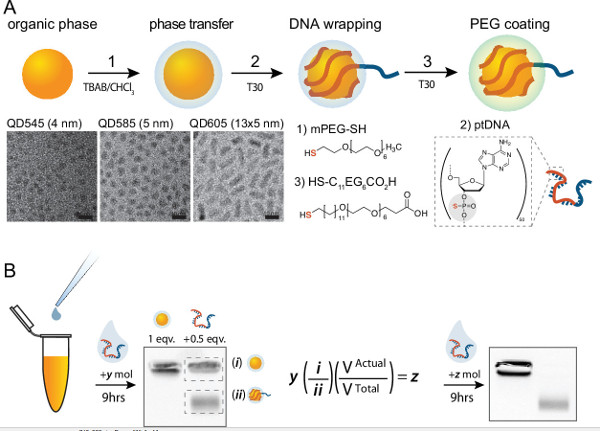
Figure 2. Scheme for production of mQDs. (A) Organic-phase QDs are transferred to the aqueous phase by treatment with mPEG thiol and TBAB, wrapped with ptDNA, and passivated with carboxy PEG6 alkane thiol. Representative TEM images are organic QD545, QD585, and QD605, respectively. (B) A method for empirically determining the stoichiometry of interaction between QDs and ptDNA in step two above. QD & ptDNA stoichiometries are empirically verified by densitometry such that the final reaction stoichiometry is 1:1 (QD:ptDNA). The initial number of moles of ptDNA is multiplied by the ratio of QD:mQD, and adjusted by the actual volume after conjugation to yield the number of moles required to achieve 1:1 conjugation. Please click here to view a larger version of this figure.
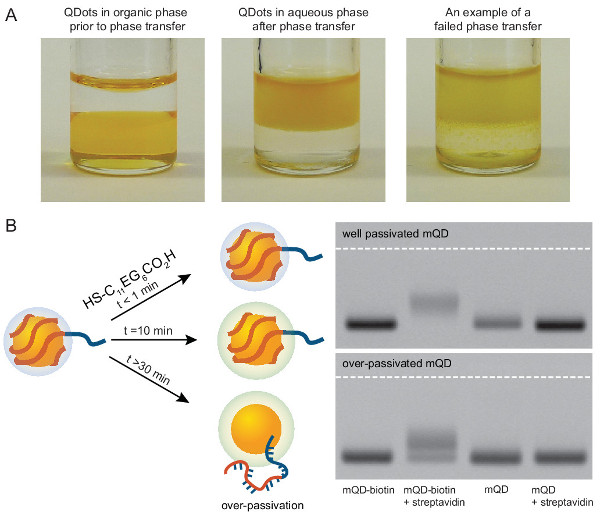
Figure 3. Experimental details for production of mQDs. (A) Representative photos of successful and failed phase transfer. QDs should visually transfer between the dense organic phase and the less-dense aqueous phase. Incomplete phase separation indicates poor transfer. (B) ptDNA can be replaced by the alkane-PEG-thiol. Right gel data is representative of a reaction where a significant fraction of ptDNA have been displaced from the QDs due to over-passivation. Please click here to view a larger version of this figure.
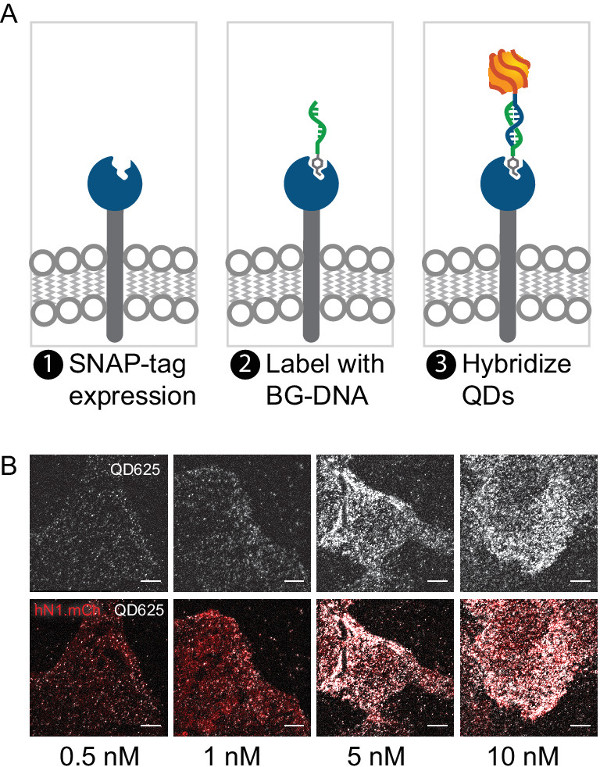
Figure 4. Labeling SNAP-tagged proteins on live cells. (A) Schematic demonstrating the expression, attachment and labeling of a SNAP-tagged receptor with a BG-targeted mQD. (B) Representative labeling of cells expressing a SNAP-Notch-mCherry construct at various mQD concentrations. mQDs passivated with PEG12 colocalize with mCherry indicating specific labeling. Lower labeling densities (<0.5 nM) are generally preferable for single particle tracking. Scale bar is 10 μm. Please click here to view a larger version of this figure.
Discussion
The modularity of the mQD design enables an increased degree of experimental flexibility. For example, a variety of mQDs can be quickly prepared in unique colors allowing for the simultaneous imaging of multiple targets. The ssDNA targeting sequence can direct mQDs to proteins, sugars12, lipids and surfaces13. A number of enzymatic tags are available with orthogonal reactivities, allowing multiple targets to be imaged simultaneously with differentially targeted mQDs. In addition to targeting with the SNAP tag, labeling of target proteins with mQDs using the CLIP tag, the HALO tag, and biotinylated proteins was also successful. This protocol demonstrates the specific labeling of a surface receptor on live cells with these mQDs, but the protocol could easily be adapted to a number of different contexts.
The significant methodological insight in producing mQDs with defined valency is that ~50 phosphorothioate-linked bases are required to wrap the QDs (and thus prevent two molecules from binding simultaneously). A poly-AS50 sequence reproducibly and stably bound 605 nm QDs from Life Technologies. Although this product has been discontinued, the steric exclusion strategy is generalizable to similar products from other vendors having different sizes, shapes, spectral properties.
The efficiency and stability of ptDNA-wrapped QDs depends critically on the surface chemistry and structure of the QDs. Therefore, the success of a protocol will depend upon the commercial source and chemical structure of the QDs. For the purposes of this protocol, three major points of difference exist between various commercial sources of QDs: a difference in conditions necessary for phase transfer; a difference in the strength of initial PEG-thiol-ligand binding, possibly hindering displacement by the ptDNA; and a difference in the amount of exposed CdSe core, which can lead to the quenching of the QD by the mPEG thiol phase transfer conditions.
As of publication, QDs with the best structure for production of mQDs are 4–10 nm CdSe/ZnS core/shell QDs purchased from Life Technologies with emission spectra at 545, 585, 605 & 625 nm (Figure 2A). QDs based upon the ‘Vivid’ formulation (545, 605, etc.) quench upon addition of mPEG thiol and are not suitable for this application. QDs from Aldrich and Ocean Nanotech work well, but require longer phase transfer steps and pretreatment with trioctylphosphine oxide. This protocol has been optimized for QDs from Life Technologies.
The poly-A phosphorothioate sequence used to wrap the QDs is terminated with a native 20-mer DNA tail containing the sequence of (ACTG)5 to which a targeting strand may hybridize. This sequence is convenient, as it has little to no secondary structure, and will remain hybridized at 37 °C in PBS. If there is access to a DNA synthesizer, the ptDNA can by synthesized and then purified by reverse phase high performance liquid chromatography (HPLC) using a C8 column. The ptDNA will elute later on the HPLC than equivalent oligonucleotides with a native backbone. We typically leave the 5’ DMT protecting group on our phosphorothioate oligonucleotides after purification.
Passivation of the ptDNA-wrapped QDs is usually required in order to improve colloidal stability of QDs and reduce background binding for most experimental applications. The protocol uses a PEG-layer to passivate the QDs. Carboxy PEG alkane thiol with additional PEG units ((CO2H)CH2O(CH2CH2O)12C11H23SH, carboxy-PEG12 alkane thiol) provides significantly reduced background, though the longer PEGs are both larger, and generally more expensive. mQDs coated with carboxy PEG alkane thiol ligands are highly stable in physiological buffers such as phosphate buffered salines and culture media. Long-term storage (> 8 months) of mQDs at 4 °C showed no significant aggregation or ptDNA detachment11. Depending upon the experiment, PEG passivation of the QDs alone does not always sufficiently reduce non-specific binding of the mQDs. Incubating both cells and mQDs in phosphate buffered saline (PBS) containing 3% BSA for 20 min prior to use substantially reduces non-specific binding to cells, though it does increase the apparent hydrodynamic radius of the mQDs by ~50%. Passivation with 0.5% casein reduces the non-specific binding even further but it increases the apparent size to a greater extent than BSA.
The 5’ end of a DNA strand complementary to the mQDs can be modified to enable targeting of a number of different biomolecules. There are a number of established techniques available to covalently modify proteins, lipids & sugars with single stranded DNA (ssDNA). So long as the ssDNA is presented extracellularly, it is accessible to soluble mQDs. mQDs with the above sequences will rapidly hybridize with their complementary DNA strand under cell culture conditions. A 10–20x poly-(CT) spacer between the ptDNA and the targeting sequence may be required for efficient targeting so as to elevate the binding sequence above the thick and negatively charged glycocalyx of the cell. For this protocol we chose to produce a BG-DNA with a complementary sequence of (CAGT)5 that will both hybridized to the mQDs and covalently link itself to a SNAP-tag protein for rapid and specific labeling. A similar protocol functions well for coupling other NHS-esters to amino-modified oligonucleotides.
For single-molecule imaging, a low density of labeling is typically required to resolve individual molecules. A final mQD concentration of ~0.5 nM in PBS with passivating agent is a good target. However, these high dilutions sometimes resulted in under-labeling of the cells. If this occurs, additional mQD can be added until an optimal density of labeling is observed. In the case of SNAP-tagged human Notch1, concentrations of >10 nM mQD produced dense labeling cells while 0.5 nM mQD resulted in the attachment of ~20 mQDs at the basal surface of targeted cell (see Figure 4B). Cell labeling with mQDs was highly dependent of the confluency of plated cells. Overly confluent cells do not label at their basal surfaces.
In summary, a simple method to generate monovalent and modular QDs was described. These mQDs find utility in wide range of live cell imaging applications, as demonstrated by imaging the Notch receptor on live U2OS cells. Applicability of mQDs is not limited to this specific case, but can be potentially extended for other cellular targets such as other proteins, nucleic acids, and enzymes.
Offenlegungen
The authors have nothing to disclose.
Acknowledgements
Funding provided by DOD W81XWH-10-1-1023 (Z.J.G.), grant P50 GM081879 from the UCSF Center for Systems and Synthetic Biology (Z.J.G.), NIH 5R21EB015088-02 (Y.J.) and NIH 1R21EB018044 (Z.J.G. & Y.J.). D.S. was supported by Human Frontier Science Program Cross- disciplinary postdoc research fellowship.
Materials
| Name of Material/ Equipment | Company | Catalog Number | Comments/Description |
| mQD Production: | |||
| Phosphorothioate DNA: | |||
| (A*)x50-(ACTG)x5 | IDT | N/A | Most DNA Synthesis companies |
| Quantum Dots: | |||
| QDots 525, 585, 605 & 625 | Invitrogen | Q21791MP (545), Q21711MP (585), Q21701MP (605) | Custom QD synthesis for QD625. |
| QD610 | Ocean Nanotech | QSP-610-10 | |
| QD 610 lamda | Aldrich | 731854 | |
| Chloroform, 99.8% | ACROS | 67-66-3 | |
| Tetrabutylammonium bromide, 98.0% | Sigma-Aldrich | 426288 | |
| mPEG thiol [2,5,8,11,14,17,20-Heptaoxadocosane-22-thiol], MW 354.5, 95% | Polypure | 11156-0695 | |
| HSC11EG6CO2H [HS-(CH2)11-(OCH2CH2)6-OCH2CO2H] | ProChimia | TH 003-m11.n6-0.1 | |
| Boric Acid, 99.5% | Sigma-Aldrich | B0394 | |
| Sodium Hydroxide, 99.0% | ACROS | S/4845 | |
| Sodium Chloride, 98% | Sigma-Aldrich | 310166 | |
| Agarose LE | U.S. Biotech Sources | G02PD-125 | |
| Ethanol | Sigma-Aldrich | 459828 | |
| BG-DNA Production: | |||
| Amine DNA: | |||
| (CAGT)5-NH2 | IDT | N/A | Most DNA Synthesis companies |
| (CAGT)5(T)40-NH2 | IDT | N/A | Most DNA Synthesis companies |
| NHS-GLA-Benzylguanine | New England Biosciences | S9151 | |
| DMSO | Sigma-Aldrich | D8428 | |
| HEPES Buffer | Sigma-Aldrich | 83264 | |
| NAP5 Column | GE Healthcare | 17-0853-01 | |
| C18 Column | |||
| Acetonitrile | |||
| Mammalian Cell Culture & Imaging: | |||
| Cell line expressing SNAP-tagged protein | New England Biosciences | E9100S | |
| McCoys 5A | UCSF Cell Culture Facility (?) | Specific to U2OS culture | |
| Fetal Bovine Serum | UCSF Cell Culture Facility (?) | Specific to U2OS culture | |
| PBS | UCSF Cell Culture Facility (?) | ||
| Nunc Lab-tek II Chambered Coverglass | Thermo-Fischer Scientific | 155409 | |
| Matriplate 96-well plate | Brooks Life Science Systems | MGB096-1-2-LG-L | |
| BSA | |||
| 5% Alkali-soluble Casein | EMD Millipore | 70955 | Not all caseins are the same |
| (Optional) DNA Synthesis Reagents: | |||
| 5’ Amine modifier C6 | Glen Research | Oct-06 | |
| dA-Thiophosphoramidite | Glen Research | 10-700 | |
| Analysis Software: | |||
| FIJI | http://valelab.ucsf.edu/~schindelin/ | ||
| RyTrack.pro | http://sun.iwu.edu/~gspaldin/rytrack.html | ||
| Tracker | http://www.gartnerlab.ucsf.edu/more/software | ||
Referenzen
- Sako, Y., Minoghchi, S., Yanagida, T. Single-molecule imaging of EGFR signaling on the surface of living cells. Nature Cell Biology. 2 (3), 168-172 (2000).
- Luo, W., He, K., Xia, T., Fang, X. Single-molecule monitoring in living cells by use of fluorescence microscopy. Analytical and Bioanalytical Chemistry. 405 (1), 43-49 (2013).
- Chung, I., et al. Spatial control of EGF receptor activation by reversible dimerization on living cells. Nature. 464 (7289), 783-787 (2010).
- Fernández-Suárez, M., Ting, A. Fluorescent probes for super-resolution imaging in living cells. Nature Reviews Molecular Cell Biology. 9 (12), 929-943 (2008).
- Sark, W. G. J. H. M., Frederix, P. L. T. M., Vanden Heuvel, D. J., Gerritsen, H. C. Photooxidation and photobleaching of single CdSe/ZnS quantum dots probed by room-temperature time-resolved spectroscopy. The Journal of Physical Chemistry B. 105 (35), 8281-8284 (2001).
- Pinaud, F., Clarke, S., Sittner, A., Dahan, M. Probing cellular events, one quantum dot at a time. Nature Methods. 7 (4), 275-285 (2010).
- Michalet, X., et al. Quantum dots for live cells, in vivo imaging, and diagnostics. Science. 307 (5709), 538-544 (2005).
- Howarth, M., et al. Monovalent, reduced-size quantum dots for imaging receptors on living cells. Nature Methods. 5 (5), 397-399 (2008).
- Iyer, G., et al. Aromatic aldehyde and hydrazine activated peptide coated quantum dots for easy bioconjugation and live cell imaging. Bioconjugate Chemistry. 22 (6), 1006-1011 (2011).
- Tikhomirov, G., et al. DNA-based programming of quantum dot valency, self-assembly and luminescence. Nature Nanotechnology. 6 (8), 485-490 (2011).
- Farlow, J., Seo, D., Broaders, K. E., Taylor, M. J., Gartner, Z. J., Jun, Y. W. Formation of targeted monovalent quantum dots by steric exclusion. Nature Methods. 10 (12), 1203-1205 (2013).
- Gartner, Z. J., Bertozzi, C. R. Programmed assembly of 3-dimensional microtissues with defined cellular connectivity. Proceeding of the National Academy of Sciences USA. 106 (12), 4606-4610 (2009).
- Selden, N. S., et al. Chemically programmed cell adhesion with membrane-anchored oligonucleotides. Journal of the American Chemical Society. 134 (2), 765-768 (2012).
