The entire sample preparation technique was performed as described above and the most important and/or relevant aspects are presented below. Hydrophilic and hydrophobic internal standards were spiked into pooled plasma samples to perform direct comparisons of the internal standards and endogenous metabolite abundances using various extraction methods. Liquid chromatography-mass spectrometry (LC-MS) data was analyzed using qualitative and quantitative software and resulted in excellent recovery and separation of both the endogenous compounds and internal standards. Figure 1 demonstrates the effectiveness of the MTBE-SPE method in extracting both lipid standards (A) and endogenous compounds (B).
Overall, better extraction and coverage of the metabolites were obtained compared to other methods such as methanol extraction, or ‘MTBE only’ extraction when the number of features was compared using qualitative and quantitative software following LC-MS analysis. For example, using only methanol extraction, the variation for creatinine-D3 was 15.2%. However, with MTBE LLE, this was reduced to 1.04% CV. Using MTBE, the reproducibility of lipids and aqueous compounds were <8% and <5% respectively, compared to a simpler methanol extraction which resulted in larger variation of 29% and 15% respectively for lipids and aqueous compounds. The internal standards used to monitor lipid recoveries – testosterone-D2, C17 ceramide, 15:0 PC, and 17:0 PE increased by 26%, 200%, 100%, 400% respectively compared to using methanol alone. Similar increases were detected for fatty acid internal standards and phosphotidylcholine and phosphotidylethanolamine endogenous metabolites. Other endogenous metabolites such as sphingosines, ceramides, diacylglycerols, triacylglycerols, cholesterol, and sphingomyelin were either not detected using methanol or were detected at negligible levels. However these endogenous lipids were easily detected using MTBE extraction.
In our analysis comparing standard protocols, the following results were obtained: Methanol precipitation alone resulted in 1,851 metabolites, methanol-ethanol precipitation gave 2,073 metabolites, MTBE with liquid-liquid extraction gave 3,125, and MTBE with liquid-liquid and solid-phase extraction recovered 3,806 metabolites. Therefore this approach results in a greater number of metabolites being extracted, most likely due to reduced ion suppression and cleaner samples prior to LC-MS.
Figure 2 demonstrates the efficiency in separating the hydrophobic metabolites into their respective chemical classes for more confident metabolite identification. There is minimal overlap of the compounds identified in the three lipid fractions following SPE. In support, Figure 3 shows the recovery of the internal standards demonstrating that ISTD’s were eluted in the fraction related to their chemical class.
Quality control samples are used to evaluate the quality of the sample preparation, to determine any batch effects when multiple days of analysis are required for a large sample set, and to monitor instrument reproducibility. Chromatograms are examined to ensure that spiked-in standards are greater than 90% recovered with mass error of less than ± 3 ppm and retention time window of less than ±5%. If these criteria are not met, the results are discarded and the samples are reanalyzed. In a case of batch effects whereby a shift in retention is observed for one batch, the data analysis software can correct for this. A previously prepared batch of pooled plasma samples underwent sample preparation. The fractions were then sub-aliquoted into autosampler vials and stored at -80 °C for use in monitoring instrument conditions throughout every sample analysis. Table 1 shows the results from these spike-in standards. The fatty acid negative ionization mode fraction (data not shown in table) was not used for analysis because the % CV of the spike-in standards for the QC sample was greater than 10%. The dataset for that fraction was therefore discarded and the instrument inspected and maintained. Table 2 shows the results from endogenous metabolites in the samples following three different days of sample preparation and triplicate instrument injections. The endogenous metabolites in the sample preparation QC samples are all reproducible, signifying the strength of the sample preparation as well as the instrument injection reproducibility.
When sample preparation steps are not properly followed, however, unreliable and inconsistent results are obtained. Figure 4 shows the results when the protein precipitation step of the method is not followed as outlined. Three operators, A, B, and C performed the same sample preparation procedure on pooled plasma samples. Operator A, rather than pipetting the required amount of supernatant per the experimental protocol, instead pipetted >1 ml for both washes with some of the pellet. This not only resulted in a higher number of false positives for that fraction, but increased the variability of the data.
The chromatographic reproducibility of the data can be seen in Figure 7. Pooled plasma samples were prepared in triplicate on separate days using protein precipitation, liquid-liquid extraction, and solid-phase extraction as described in this protocol. Each fraction was analyzed using the chromatographic separation described in section 7 of the protocol. Samples were then injected in triplicate on the LC-MS to evaluate instrument and sample preparation reproducibility. This consistent overlap demonstrates both the strength of the reproducibility of the sample preparation when prepared on three different days, as well as the strength of the chromatographic method in producing reproducible results. An increase in chemical noise is observed for the negative ionization mode of the fatty acid fraction. This may occur due to contaminants in the LC-MS solvents and can result in inconsistent quantitative metabolomic results. Therefore only metabolites which eluted prior to 9 min were analyzed.
When running long worklists, a loss of instrument sensitivity and change in buffer concentrations can occur over time resulting in decreased signal intensity and retention time shift. If the retention time overlap variation is less than 5% and the signal intensity variation is less than 10%, the data is still within standard laboratory limits. Analysis software can be used to align and normalize the data to correct for instrument and retention time drift. However, if the variation is large, then the reason has to be determined. Once this is rectified, the samples can be re-analyzed.
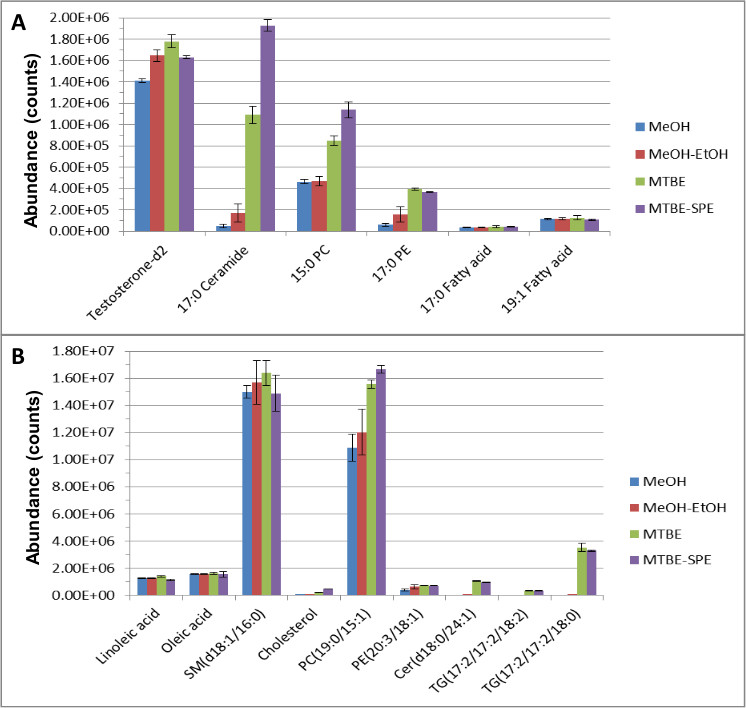
Figure 1. The abundance of lipid ISTDs (A) and endogenous metabolites (B) following extraction and reversed-phase chromatography (RPC)12. Extraction was performed and resulting samples were separated using RPC and analyzed using LC-MS in positive and negative ionization mode. This figure has been modified from Yang et al, Journal of Chromatography A 1300, 217-226 (2013).
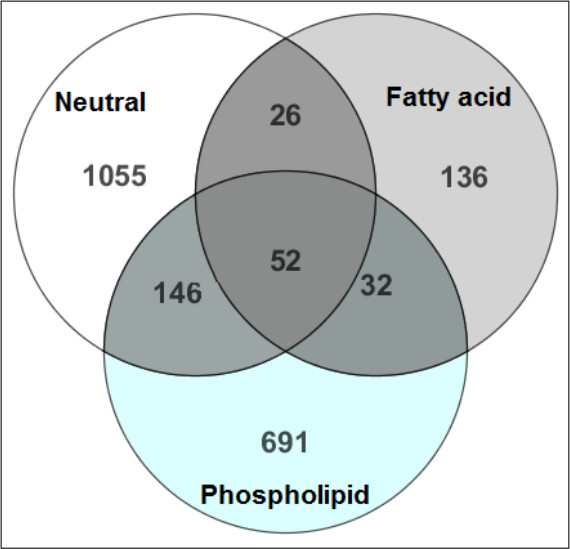
Figure 2. A comparison of MTBE-SPE fractions12. The metabolites identified in each fraction were compared to identify the amount of overlap during the SPE portion of the prep. The numbers in the Venn diagram reflect the number of metabolites detected in each fraction. Here minor overlap is observed among the three fractions, representing successful compound extraction and metabolite class separation during the SPE step. This figure has been modified from Yang et al, Journal of Chromatography A 1300, 217-226 (2013).
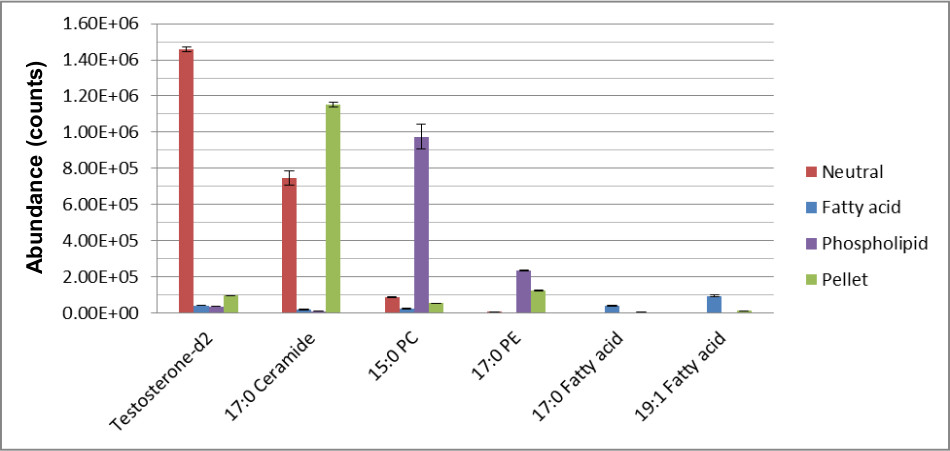
Figure 3. The recovery of ISTDs in fractions using the MTBE-SPE method12. Extraction was performed and resulting samples were separated using RPC and analyzed using LCMS in positive and negative mode as described in text. This figure has been modified from Yang et al, Journal of Chromatography A 1300, 217-226 (2013).
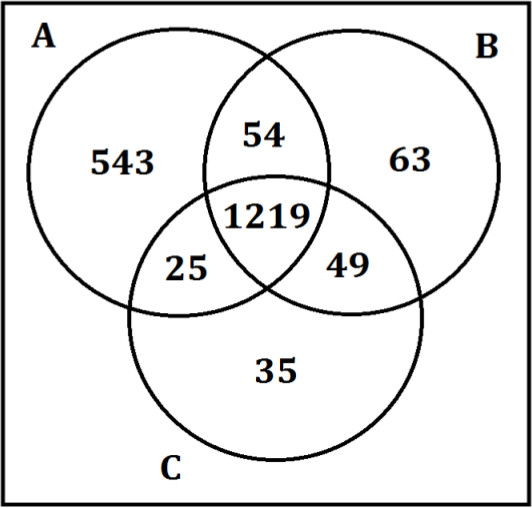
Figure 4. Results from the pellet fraction prepared by three operators. Three sample preparation operators A, B, and C performed the same protein preparation step on pooled plasma samples. The numbers in the Venn diagram reflect the number of metabolites detected by each operator. Operators B and C pipetted the required volume per the sample preparation protocol while operator A pipetted the entire supernatant and some of the pellet, resulting in over 500 more metabolites, the majority being false positives for that specific fraction.

Figure 5. Formation of protein pellet during protein precipitation step. (A) 100 µl of human plasma prior to sample preparation; (B) plasma after addition of ice cold methanol; (C) protein pellet formed on bottom of tube after centrifuging at 0 °C for 15 min at 18,000 x g.

Figure 6. Separation of the hydrophilic and hydrophobic layers during liquid-liquid extraction (LLE) step. The organic solvent methyl tert-butyl ether (MTBE) and water were used to separate the hydrophilic and hydrophobic metabolites. The MTBE layer has dissolved non-polar compounds and the water layer has dissolved polar compounds. (A) plasma supernatant after protein removal; (B) plasma after drying under nitrogen; (C) plasma after addition of MTBE; (D) addition of water to plasma and MTBE; (E) MTBE-water layer formed after centrifuging; (F) removal of top MTBE layer; (G) mainly hydrophilic layer remaining after MTBE removal.
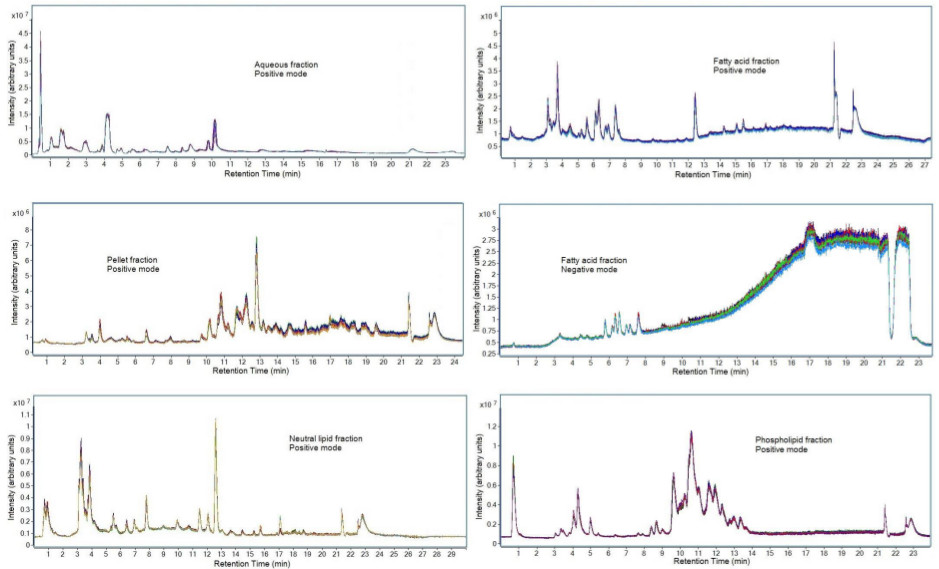
Figure 7. Chromatograms of fractions from a selected dataset. Sample preparation was performed on three separate pooled plasma QC samples and each sample was injected in triplicate on the LC-MS instrument. Represented are total ion chromatograms of the plasma samples acquired using the LC-MS parameters indicated in section 7 of the method protocol. The chromatographic representation of other biological fluids will vary due to differences in metabolite composition. Please click here to view a larger version of this figure.
| Fraction | Ionization Mode | Internal Standard | n | Average Peak Area | Peak Area % CV |
| Aqueous | Positive | Creatinine-D3 | 31 | 2217311 | 3.8% |
| Neutral lipid | Positive | Triglyceride-D5 | 31 | 4837032 | 9.9% |
| C17 Ceramide | 31 | 12736707 | 7.9% | ||
| Phospholipid | Positive | 15:0 PC | 32 | 1248929 | 9.3% |
| 17:0 PE | 32 | 517234 | 7.9% |
Table 1. Quality control results from spiked internal standards. Pooled plasma samples from an emphysema mouse model dataset were analyzed to monitor instrument conditions on a daily basis for this multi-week study. Quantitative analysis software was used to determine the peak areas of the internal standards, (n = number of instrument QC injections).
| Fraction | Ionization Mode | Endogenous metabolites | Average Peak Area | Peak Area % CV |
| Aqueous | Positive | Creatinine | 2554574 | 2.3% |
| Valine | 3712151 | 3.3% | ||
| Glucose | 2669190 | 6.9% | ||
| Neutral lipid | Positive | 3-Dehydrosphinganine | 226644 | 3.9% |
| DG(16:0/16:1/0:0) | 11301 | 8.2% | ||
| DG(P-14:0/18:1) | 364119 | 1.9% | ||
| Phospholipid | Positive | PC(24:0/0:0) | 27599 | 0.9% |
| PC16:0/22:6) | 2873326 | 4.5% | ||
| PI(16:0/18:1) | 112998 | 4.4% | ||
| Fatty acid | Positive | 10-oxo-5,8-decadienoic acid | 1363284 | 2.3% |
| 16-oxo-heptadecanoic acid | 83700 | 2.9% | ||
| 2-methyl valeric acid | 285782 | 5.7% | ||
| Fatty acid | Negative | 10-hydroxy-8-octadecenoic acid | 10042 | 4.9% |
| (R)-laballenic acid | 173929 | 6.5% | ||
| 2-keto valeric acid | 35488 | 6.0% |
Table 2. Quality control results from endogenous metabolites. Pooled plasma samples from a human disease dataset were analyzed to monitor sample preparation reproducibility on separate days. Samples were prepared in triplicate over three days, (n = 9 prep QC injections).
