As part of an ongoing study of copper-binding protein models, the structure of the conserved sequence MT/HCXXC within the linear peptide MTCSGCSRPG was determined by solution state NMR. The peptide was reacted with CuCl under inert environment and the pH was measured as ~3.0 by a universal stick. The amide region of the peptide showed an expansion upon reaction with copper, from 6.7-8.5 to 6.6-9.0 ppm (Figure 2). Line broadening due to slight copper oxidation is evident in the baseline.
The copper-reacted peptide sample was stable with time (Figure 7) and the spectra were well-resolved (Figure 8) and gave 81 NOE interactions that were acquired by ROESY experiment since the molecule gave near zero NOE interactions in the NOESY spectrum (see theoretical explanation in Figure 6).
The ensemble of the peptide derived for the reacted sample, but using no constraints to the metal gave 47 out of 50 nonviolated structures with an RMSD value of 1.44 and 2.07 Å on the backbone and heavy atoms, respectively. Of these, 13 low-energy conformers were chosen for further analysis, with RMSD values of 0.25 and 0.61 Å on the backbone and heavy atoms, respectively.
The local RMSD plot (Figure 15) showed a region of stability between residues 3 and 7, in addition to the rigid C-terminal region including a proline residue. This region is found in a bend conformation between residues 4 and 7 in all conformations (Figure 19), which are stabilized by hydrogen bonding between backbone donors and acceptors, Gly5 and Thr2; Cys6 and Cys3 (Figure 20). This bend is also evident between C3 and S7 by the reduced 3JHNHα values in this region (Figure 11).
The conformations were superimposed over this region (Figure 16) and analyzed for possible binding residues. When considering Cys3, Cys6 and Met1 as potential binding residues, the shortest S.”.S distance was that between the thiolato groups of Cys3 and Cys6 (7.9±0.1 Å relative to 9.1±1.1 and 9.4±0.9 Å for SMet1…SCys3 and SMet1…SCys6, respectively). Copper-binding was introduced and the calculation was repeated to give the ensemble used for analysis (Figure 17).
The low-energy ensemble of the copper-bound peptide shows that the N-terminal amine is proximate to the bound copper (3.5-5.5 Å).
The electrostatic potential distribution on the surface of the molecule showed that arginine residue extends from the backbone of the peptide, forming a positive lobe of electrostatic potential, whereas the backbone carbonyls are arranged in a line forming a less prominent-1 kT/e electrostatic potential (Figure 23).
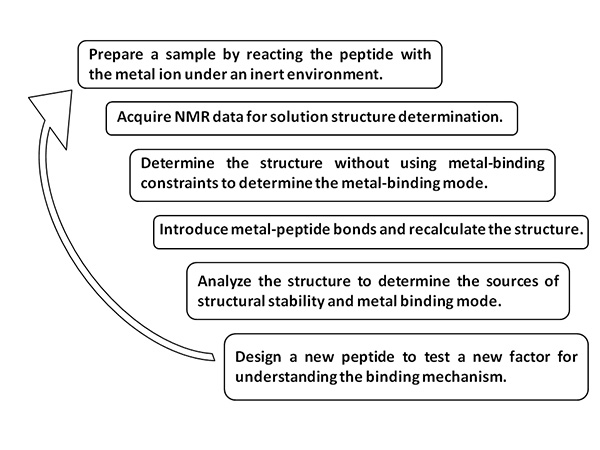
Figure 1. Scheme of the method.Click here to view larger image.
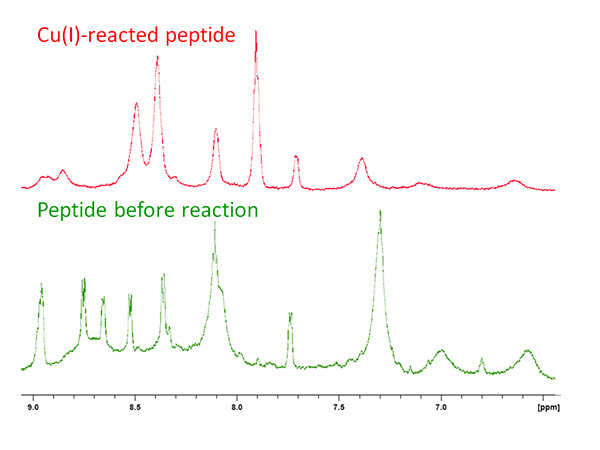
Figure 2. 1D NMR spectrum of apo- (green) and copper-reacted peptide (red) showing the change in distribution of amide resonances.Click here to view larger image.
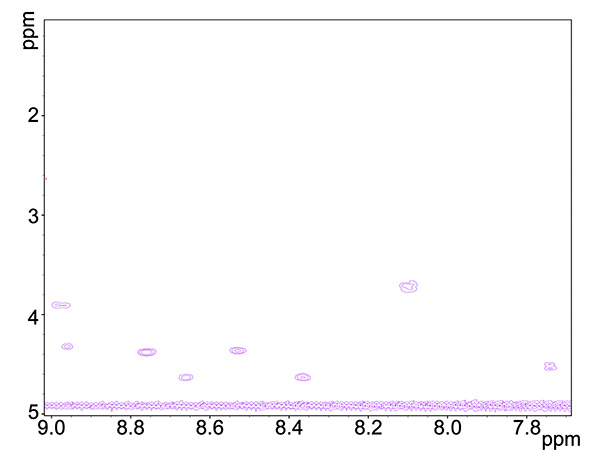
Figure 3. COSY spectrum of copper-reacted peptide, showing HN-Ha neighboring proton group interactions.Click here to view larger image.
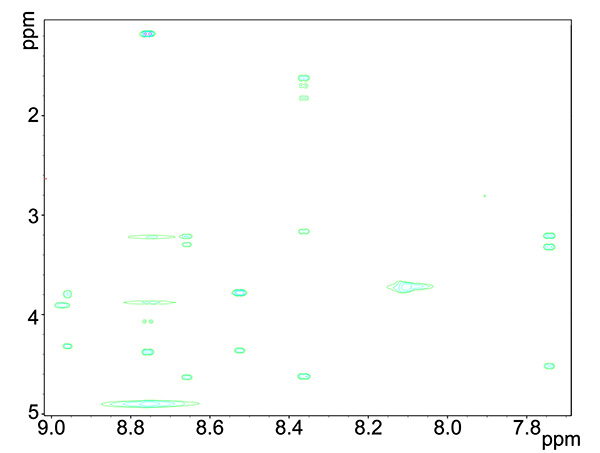
Figure 4. TOCSY spectrum of copper-reacted peptide, showing HN interactions with proton groups within the residue.Click here to view larger image.
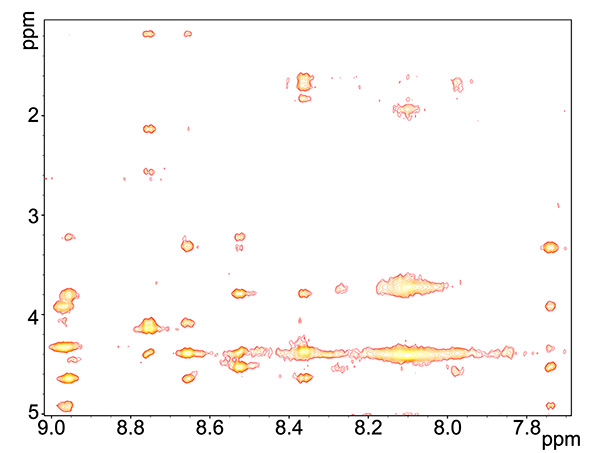
Figure 5. ROESY spectrum of copper-reacted peptide, showing HN interactions with all proximate proton groups, independent of being covalently bound to amide.Click here to view larger image.
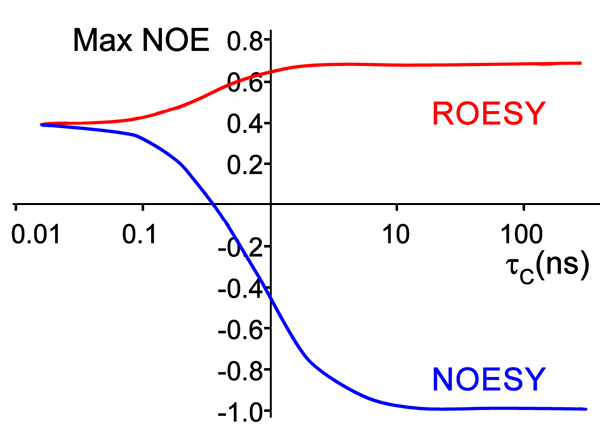
Figure 6. Theoretical calculation of NOE intensity of molecule by NOESY and ROESY experiment versus the correlation time, indicative of the rotation rate, which is dependent on effective size and spectrometer field15.Click here to view larger image.
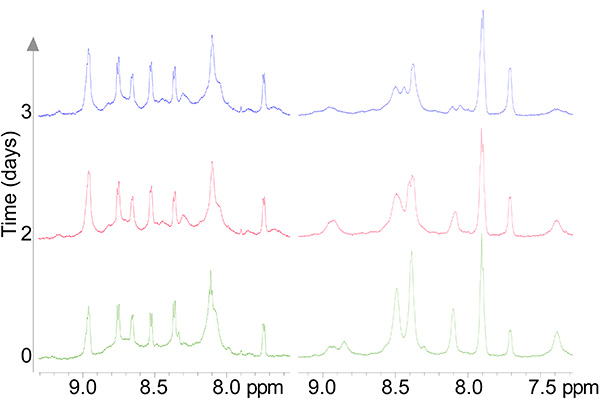
Figure 7. 1D spectra of a stable copper complex (left pane) and the apo peptide (right pane) undergoing oxidation as a function of time.Click here to view larger image.
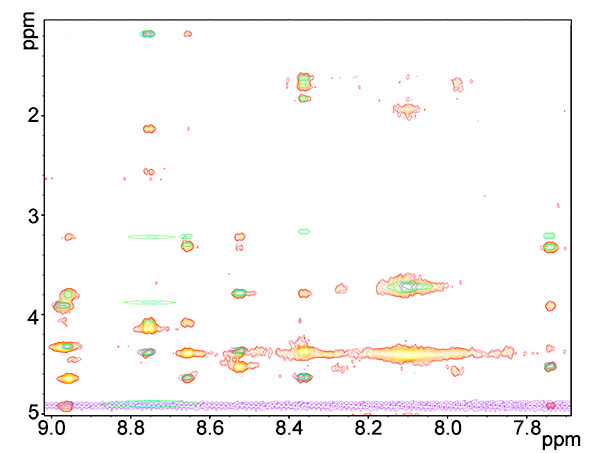
Figure 8. Overlay of fingerprint regions of ROESY (red-yellows), TOCSY (blue-greens) and COSY (purple) spectra of copper-bound peptide.Click here to view larger image.
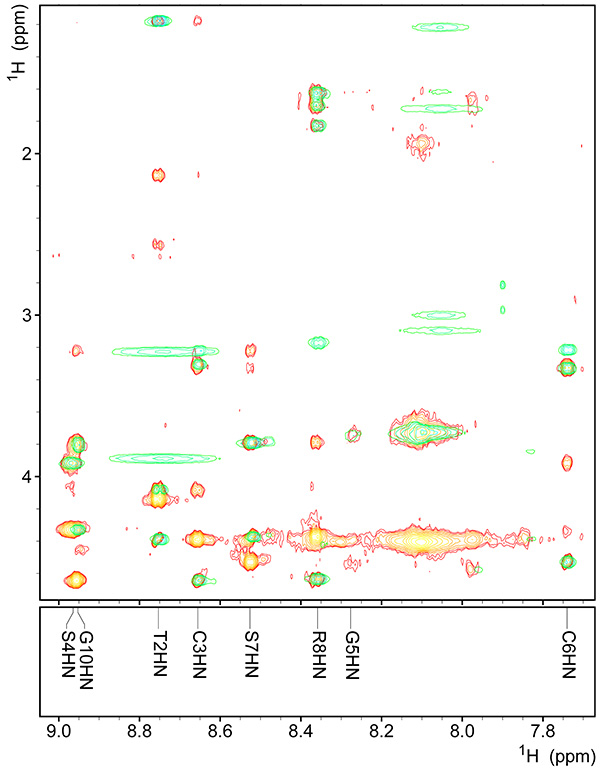
Figure 9. Assigned fingerprint region of spectrum of copper-bound peptide.Click here to view larger image.
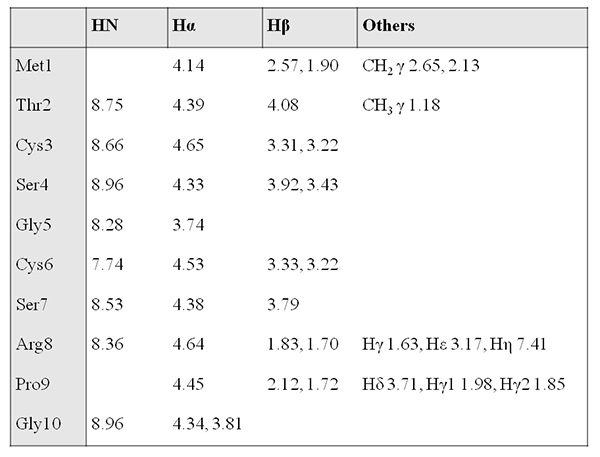
Figure 10. 1H chemical shift assignment table (ppm).Click here to view larger image.
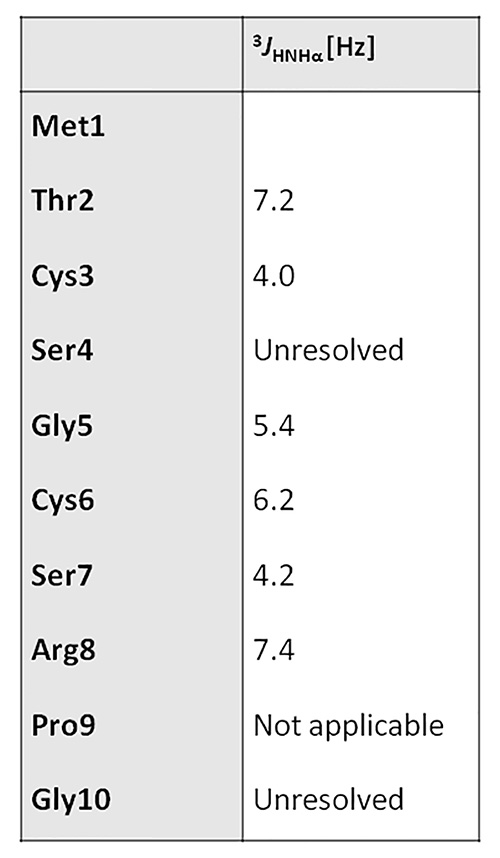
Figure 11. 3JHNHα values table.Click here to view larger image.

Figure 12 . Inter-residual constrains list.Click here to view larger image.

Figure 13. Statistics on NMR-derived NOE constraint file used to generate ensemble of peptide.Click here to view larger image.
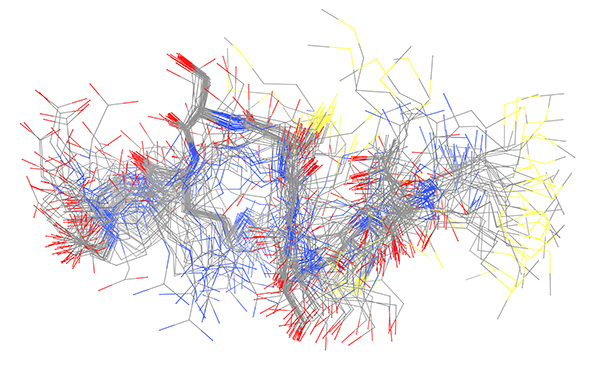
Figure 14. Ensemble derived from NMR data of copper-bound peptide. Complete 50-member ensemble representing all samples conformations superimposed on backbone.
Click here to view larger image.
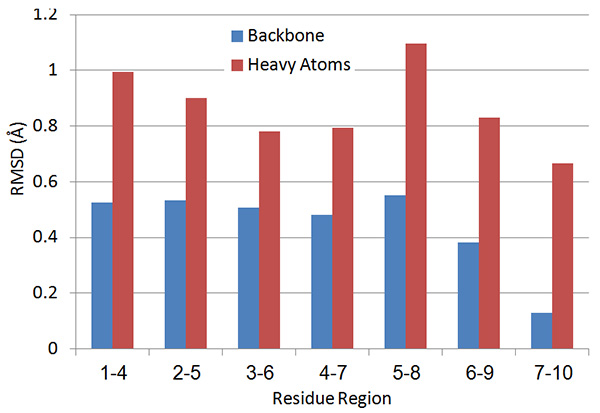
Figure 15. Local four-residue RMSD values along the peptide sequence.Click here to view larger image.
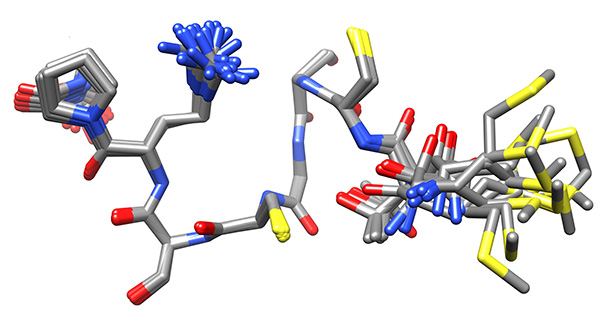
Figure 16. Low-energy ensemble of copper-bound peptide without any restraints to the metal, representing the low-energy conformations that have no violations of experimental NMR-derived constraints, superimposed on core region of larger stability. This ensemble will be used to determine the residues that interact with the copper ion.Click here to view larger image.
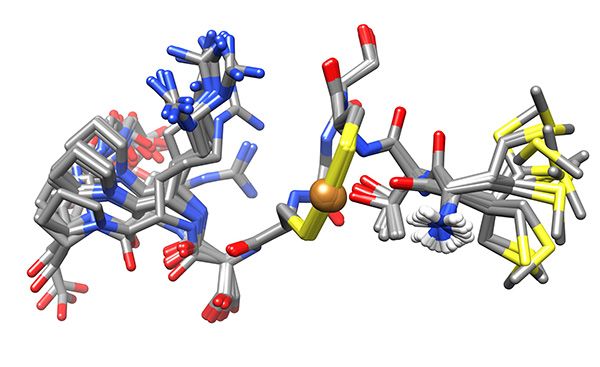
Figure 17. Low-energy ensemble of copper-bound peptide, representing the low-energy conformations that have no violations of experimental NMR-derived constraints.Click here to view larger image.
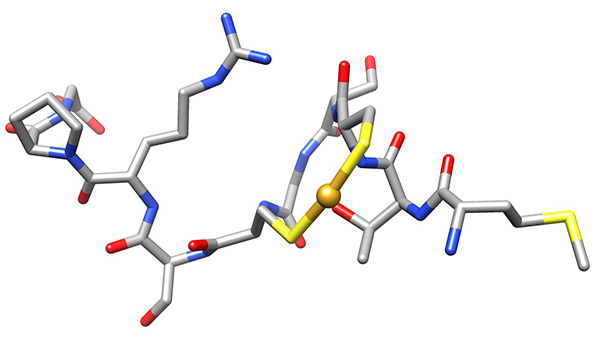
Figure 18. Low-energy conformer of copper-bound peptide, representing the low-energy conformations that have no violations of experimental NMR-derived constraints.Click here to view larger image.
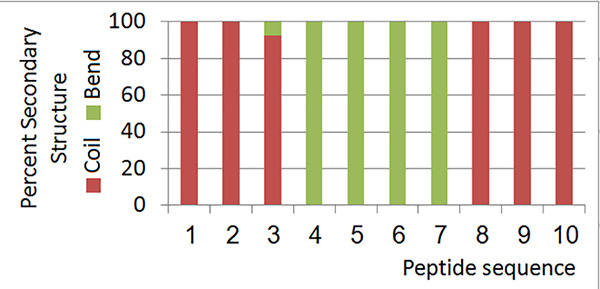
Figure 19. Secondary structure information for all members of the low-energy ensemble.Click here to view larger image.
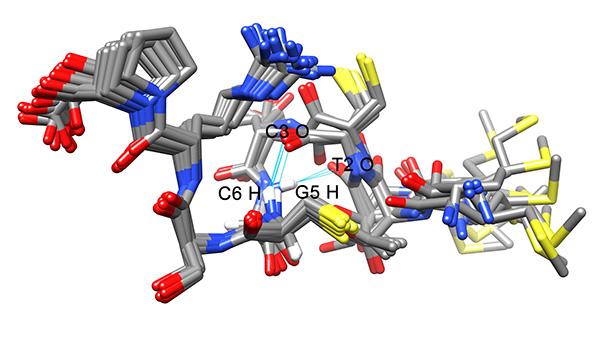
Figure 20. Intramolecular hydrogen bounds found in the conformers of the ensemble.Click here to view larger image.
Figure 21. Low-energy representative conformation in grid used to calculate electrostatic potential distribution.Click here to view larger image.
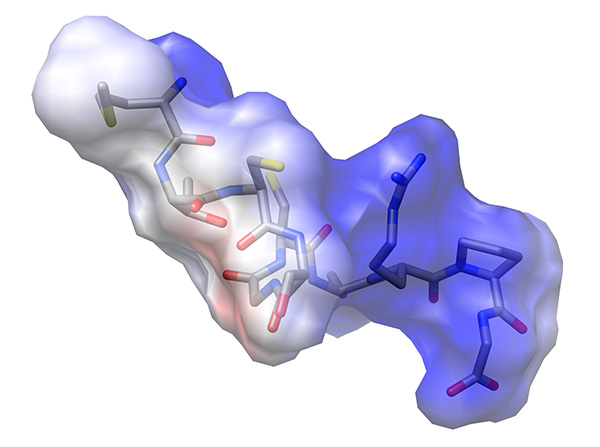
Figure 22. Electrostatic potential mapped onto Van der Waals surface of low-energy representative conformation of peptide.Click here to view larger image.
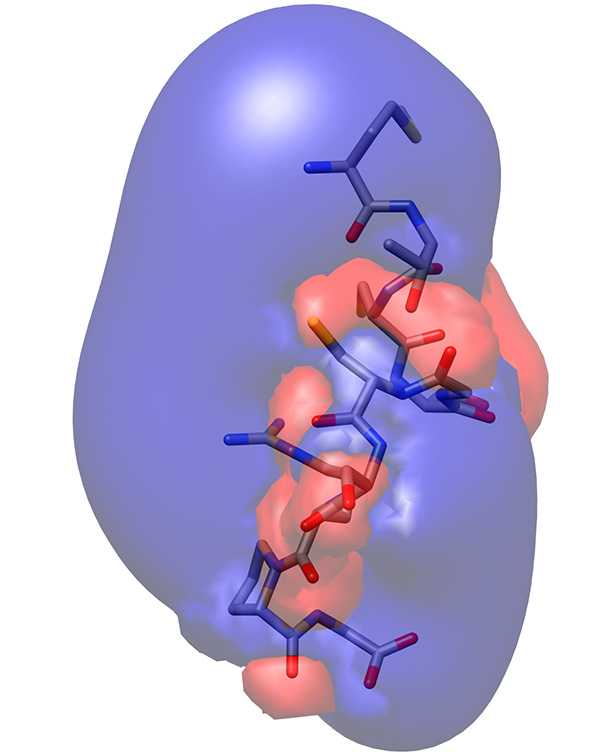
Figure 23. Electrostatic potential iso-surface of low-energy representative conformation of peptide showing +/- 1 kT/e in blue and red, respectively.Click here to view larger image.
