4D Imaging of Protein Aggregation in Live Cells
Summary
Cellular viability depends on timely and efficient management of protein misfolding. Here we describe a method for visualizing the different potential fates of a misfolded protein: refolding, degradation, or sequestration in inclusions. We demonstrate the use of a folding sensor, Ubc9ts, for monitoring proteostasis and aggregation quality control in live cells using 4D microscopy.
Abstract
One of the key tasks of any living cell is maintaining the proper folding of newly synthesized proteins in the face of ever-changing environmental conditions and an intracellular environment that is tightly packed, sticky, and hazardous to protein stability1. The ability to dynamically balance protein production, folding and degradation demands highly-specialized quality control machinery, whose absolute necessity is observed best when it malfunctions. Diseases such as ALS, Alzheimer’s, Parkinson’s, and certain forms of Cystic Fibrosis have a direct link to protein folding quality control components2, and therefore future therapeutic development requires a basic understanding of underlying processes. Our experimental challenge is to understand how cells integrate damage signals and mount responses that are tailored to diverse circumstances.
The primary reason why protein misfolding represents an existential threat to the cell is the propensity of incorrectly folded proteins to aggregate, thus causing a global perturbation of the crowded and delicate intracellular folding environment1. The folding health, or “proteostasis,” of the cellular proteome is maintained, even under the duress of aging, stress and oxidative damage, by the coordinated action of different mechanistic units in an elaborate quality control system3,4. A specialized machinery of molecular chaperones can bind non-native polypeptides and promote their folding into the native state1, target them for degradation by the ubiquitin-proteasome system5, or direct them to protective aggregation inclusions6-9.
In eukaryotes, the cytosolic aggregation quality control load is partitioned between two compartments8-10: the juxtanuclear quality control compartment (JUNQ) and the insoluble protein deposit (IPOD) (Figure 1 – model). Proteins that are ubiquitinated by the protein folding quality control machinery are delivered to the JUNQ, where they are processed for degradation by the proteasome. Misfolded proteins that are not ubiquitinated are diverted to the IPOD, where they are actively aggregated in a protective compartment.
Up until this point, the methodological paradigm of live-cell fluorescence microscopy has largely been to label proteins and track their locations in the cell at specific time-points and usually in two dimensions. As new technologies have begun to grant experimenters unprecedented access to the submicron scale in living cells, the dynamic architecture of the cytosol has come into view as a challenging new frontier for experimental characterization. We present a method for rapidly monitoring the 3D spatial distributions of multiple fluorescently labeled proteins in the yeast cytosol over time. 3D timelapse (4D imaging) is not merely a technical challenge; rather, it also facilitates a dramatic shift in the conceptual framework used to analyze cellular structure.
We utilize a cytosolic folding sensor protein in live yeast to visualize distinct fates for misfolded proteins in cellular aggregation quality control, using rapid 4D fluorescent imaging. The temperature sensitive mutant of the Ubc9 protein10-12 (Ubc9ts) is extremely effective both as a sensor of cellular proteostasis, and a physiological model for tracking aggregation quality control. As with most ts proteins, Ubc9ts is fully folded and functional at permissive temperatures due to active cellular chaperones. Above 30 °C, or when the cell faces misfolding stress, Ubc9ts misfolds and follows the fate of a native globular protein that has been misfolded due to mutation, heat denaturation, or oxidative damage. By fusing it to GFP or other fluorophores, it can be tracked in 3D as it forms Stress Foci, or is directed to JUNQ or IPOD.
Protocol
1. Yeast Preparations
- Transform yeast strains with a plasmid carrying a GAL1-GFP-Y68L (UBC9ts) cassette.
- Grow yeast in synthetic media containing 2% raffinose for 24 hr and back diluted to 2% galactose containing media for 16 hr or O/N. Incubate cells at 30 °C while shaking at 200 rpm.
- The following morning, dilute the query strain to OD600= 0.2. Shake for 4-6 hr at 30 °C (200 rpm) until the culture reaches OD600= 0.8-1.0.
- To repress expression (so as to monitor only the already-translated and folded pool of GFP-Ubc9ts), change media to synthetic media supplemented with 2% glucose (SD) 30 min prior to imaging.
- Treatment – Heat shock the cells for 20 min at 37 °C to induce misfolding, or continuously in microscope incubator to follow protein aggregation in heat-shock conditions. Note – if you are using the microscope at any temperature above RT, pre-heat for about 2 hr to equilibrate the temperature along the body of the microscope and the objectives (see below).
2. PlateSlide Preparations
- Choose appropriate plate. The main consideration is the ability to maintain focus during imaging acquisition. The following points are critical for 4D imaging and not for 2D time lapses or 3D imaging.
- Multiple wells plate- the bottom of the different wells may not be homogenous (i.e. the wells will be at different heights relative to the objective). This will make it difficult to maintain focus across x y points and time points, regardless of the autofocusing technique being used.
- Slide material: glass vs. plastic. Glass bottomed slides are more z-homogenous between wells, but are more expensive.
- Thickness of the plate’s bottom: we usually use coverslip-bottom plates index 1.5, but index 1 is also acceptable depending on the objective.
- Cover bottom of the plateslide with ConA (0.25 mg/ml) for 10 min. ConA is used to adhere cells to the slide, which enables following a single cell over time.
- Remove ConA and incubate the slide in a chemical hood so that excess ConA will evaporate.
- Plate 200 μl of yeast sample (OD600= 0.5) into the ConAed well.
- Incubate for 15 min, as to enable cells stick to the surface of the plate.
- Extract the medium, and wash three times with new medium to get one layer of cells. Note: if a long time lapse is planned, seed the cells sparsely so that new buds won’t fill and interrupt the region of interest.
3. Microscope Preparations
- We use a Nikon A1Rsi confocal microscope with a few non-standard modifications. For yeast imaging we use up to 4 lasers (405 nm, 50 mW CUBE laser; 457-514 nm, 65 mW Argon Ion laser; 561 nm, 50 mW Sapphire laser; and 640 nm, 40 mW CUBE laser), and up to 4 PMTs equipped with filters. Most of our imaging is done with a green filter set for EGFP and a red filter set for mCherry and tdTomato. With GFP and mCherry there is almost no bleedthrough, therefore we often use simultaneous scanning. However, line-scanning can also be used to prevent bleedthrough when it does occur. Our confocal is also equipped with a PInano Piezo stage (MCL), which can make 3 msec steps in z, enabling 2-3 z-stacks (30-50 sections) per second. The systems also has a spectral detector, Perfect Focus (laser offset), and two scanners – a galvano scanner and a resonant scanner.
- Choose appropriate objective. Main considerations:
- Water/Oil/Air objective: the main consideration is the refractive index of the imaging medium vs the medium of the sample.
- Oil objective advantages:
- Oil objectives can have higher numerical apertures (oil breaks light more than water, and therefore more photons go back to the objective). Oil objectives can have up to NA 1.49, compared to 1.27 for water. (However, this is not actually a huge difference in resolution).
- The refractive index of oil matches the refractive index of glass, therefore no photons are lost going from the sample, through the glass, to the objective. (Note – chose an immersion oil that has a refractive index that is appropriate for your objective and your coverslip).
- Oil enables hassle-free long time-lapses since it does not evaporate.
- The higher resolution enabled by the higher NA of oil objectives degrades rapidly if the light has to travel through an aqueous medium (such as a cell).
- Images typically have more spherical aberrations and a “stretched out” effect in 3D.
- Oil is sticky, messy, and a hazard to objectives.
- Water objective advantages
- The cell itself is aqueous, therefore there are fewer distortions in z, and the resolution remains high deep into an aqueous sample.
- Clean and user friendly. Water objective disadvantages: evaporates quickly, therefore not suitable for long movies without special arrangements being made to pump water continuously to the objective.
- Air objective advantages: 1. No material is lost/evaporates during multiple point acquisition or a long movie. 2. Clean and user friendly. Disadvantage: low resolution and low signal sensitivity.
- Oil objective advantages:
- Room and incubator temperature: changing temperature affects matter properties, and in delicate systems changes the focus. The temperature should be adjusted before choosing points for acquisition. Changing the temperature during acquisition will disrupt imaging and will result in loss of focus. We let our microscope system equilibrate for 2 hr at the desired temperature before imaging.
- Correction collars: Many objectives come with a correction ring enabling the objective to be adjusted for a given cover-slip thickness. We recommend adjusting the correction collar while using fluorescent beads to visualize the point spread function. This will ensure correct settings for every sample.
- Stable sample holders: we find that most commercially available sample holders are the weakest part of the microscope. They are often wobbly and not straight. This is a disaster for high-resolution, multi-point 4D imaging. We design our own sample holders that are straight and fit tightly onto the stage. They can also be bolted down when needed.
- Multi-point acquisition: our imaging system is equipped with the Nikon “Perfect focus” system, which is basically a laser-based auto-focus mechanism. However, autofocusing is not necessarily necessary with 4D imaging, especially if the system does not drift a lot.
- Water/Oil/Air objective: the main consideration is the refractive index of the imaging medium vs the medium of the sample.
4. Imaging
- Turn on the system: lasers, stage, controller, camera and computer software.
- Place plate on stage holder, properly secured and stabilized.
- Use eye port to determine location and orientation of yeast.
- Using brightfield, assess cell health and viability according to shape and texture.
- Turn on epifluorescent light according to the relevant fluorophore (e.g. FITC filter for GFP), and focus on cells displaying the phenotype which is subject to your research. Cells which are highly fluorescent in more than one wavelength may be dead and therefore autofluorescent. We visualize the nucleus with a tdTomato fluorophore fused to an SV40 NLS signal (NLS-TFP). TFP is twice the size of GFP, and is therefore above the diffusion limit of the nucleus, therefore it works very well as a nuclear marker. We can also excite TFP with a green (488 nm) laser simultaneously as GFP, but collect the green and red emissions into two separate PMTs for spectral resolution.
- Adjust the following settings to minimize noise and oversaturation:
- Laser power – photobleaching and phototoxicity vs. brightness of image should be considered.
- Gain: affects camera sensitivity, thus Signal to Noise ratio.
- Pinhole diameter – should be adjusted according to the laser with the shorter wavelength. The pinhole diameter determines the thickness (height) of the optical section imaged. Thus, opening the pinhole will collect more light (let more photons in), but will decrease resolution in z (less confocality).
- Useful tips:
- If different fluorophores emit congruent wavelength, use the Spectral Detector feature which enables the choice of virtual filters. Keep in mind that spectral detectors are less sensitive than regular PMTs.
- If different fluorophores are excited by the same laser, use the line scanning feature.
- A Galvano scanner enables more sensitivity, but has a higher risk for photobleaching. A Resonant scanner enables faster acquisition, thus lowering the risk for photobleaching, but is less sensitive.
- Averaging between 2 and 16 images, it enhances signal to noise ratio, but makes acquisition slower and, of course, involves more exposure of the sample to the laser.
- Duration of acquisition- depends on the biological question (e.g JUNQ and IPOD formation takes ~2 hr). We have imaged yeast with the resonant scanner for up to 30 hr in 3D time-lapse.
- Time lapse intervals- Smaller intervals will create a more coherent movie but might cause photo bleaching and therefore loss of signal.
Representative Results
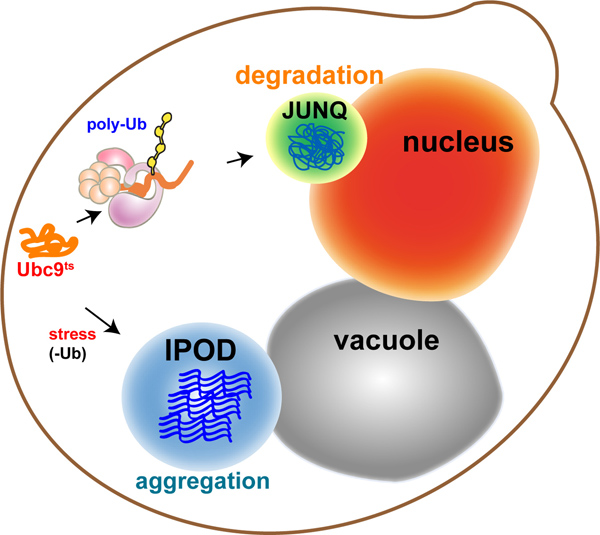
Figure 1. Model: Sub-cellular compartmentalization of misfolded proteins. Quality control machinery directs misfolded proteins into distinct compartments with distinct functions: Soluble proteins, targeted for degradation, undergo poly-ubiquitination and are sent to the Juxta-NuclearQuality control compartment (JUNQ). Insoluble proteins that can’t be ubiquitinated are sent for protective sequestration to the InsolubleProteinDeposit (IPOD), adjacent to the vacuole, where they undergo active aggregation.

Figure 2. Modeling protein misfolding with GFP-Ubc9ts. Under normal conditions, GFP-Ubc9ts (green) is natively folded, and is localized diffusely in the nucleus and the cytosol. The nucleus is labeled by NLS-TFP (red). Expression of Ubc9ts was shut off by addition of 2% glucose before imaging in all experiments. Click here to view larger figure.
- Upon temperature shift to 37 °C, GFP-Ubc9ts (green) is misfolded and forms cytosolic Stress Foci. The nucleus is labeled by NLS-TFP (red).
- Upon recovery from heat shock at 23 °C, the thermally denaturated GFP-Ubc9ts is degraded, as indicated by decreased fluorescence level.
- Upon temperature shift to 37 °C and proteasome inhibition with 80 Mm MG132, GFP-Ubc9ts is misfolded and processed into JUNQ and IPOD inclusions. The nucleus is labeled by NLS-TFP (red).
- Upon temperature shift to 37 °C and ubiquitination inhibition, GFP-Ubc9ts is misfolded and processed into the IPOD inclusion. The nucleus is labeled by NLS-TFP (red). The Ubiquitin Protease 4 (Ubp4) is overexpressed to block Ubc9ts ubiquitination.

Figure 3. Time lapse of JUNQ and IPOD formation. Upon temperature shift to 37 °C and proteasome inhibition with 80 Mm MG132, GFP-Ubc9ts Stress Foci are processed into JUNQ and IPOD inclusions. The nucleus is labeled by NLS-TFP (red). 3D images were acquired at 4 min intervals. (also see Movie 1). Click here to view larger figure.
Discussion
Our intuitions about biochemical processes derive from bench top experiments in which a well-mixed solution of reactants and products is allowed to reach equilibrium in a beaker. In such a setting, the concentration of a given chemical species may be expressed as a single number, which is the ratio of a molar quantity of molecules to a macroscopic volume. Much of what we know about protein structure and function derives from using methods that reflect the classic, bulk reaction picture: western blots, centrifugations, and spectrophotometric measurements carried out on extracts from homogenates of whole populations of cells.
As the technology we use to look at cells under magnification improves by leaps and bounds, it becomes ever clearer that the conditions in which most biochemical reactions take place in vivo bear only the slightest resemblance to those of the classic bench top scenario. Not only is the interior of the cell a densely packed environment, in which crowding effects substantially alter the activities of various reactants, it is also quite the opposite of well-mixed. This accounts for the frequent disparity between in vitro and in vivo efficiencies of a wide range of complex macromolecular reactions.
Nowhere are intuitions stemming from classical in vitro biochemical experiments more prone to mislead as in questions pertaining to the in vivo folding, misfolding, and aggregation of proteins. Whereas studies of protein chemistry in bulk reactions can treat the issue of folding for a given protein as a simple yes or no question, any attempt to track the dynamics of whole populations of macromolecules in a live cell must be sensitive to the whole distribution of possible conformational outcomes available to a polypeptide chain, and in particular to the risk of misfolding and aggregation. For example, we might examine a bulk cell lysate of an aggregating protein by western blotting, and determine that the protein is mostly insoluble and not ubiquitinated. However, in the living cell a discrete sub-population of the protein, difficult to detect when averaging over many cells, may be soluble and ubiquitinated in a particular compartment where the local concentration of the species is extremely high. The latter scenario may have more important consequences for the viability of the cell than the larger bulk sub-population. Furthermore, whereas chaperones display a variety of pleiotropic behaviors and functions in vitro, it is becoming evident that in the cell their discrete functions are spatially and temporally confined.
In the newly emerging paradigm for understanding biochemistry, concentration becomes a variable property of each specific nano-environment in the cell, and the molecular events that underlie biological processes must be assayed not only in time, but also in space. The 4D imaging approach presented here enables sensitive modeling of protein misfolding in live cells, though it can be used to study any number of other biological processes and how they are regulated in space, time, and following changes in environmental conditions. In this paper we use the Ubc9ts folding sensor, which effectively demonstrates the stages and options for dealing with the onset of protein aggregation in the cytosol. In addition to illustrating the cell biology of aggregation quality control, this approach can serve as a powerful tool for deciphering the effect of specific perturbations or genetic mutations on proteostasis (for example Ubc9ts can be used to measure protein folding stress in response to oxidation, the expression of a toxic aggregate, or mutations in the quality control pathway).
4D imaging is also essential for accurately determining protein localization or colocalization between two different proteins, and for detecting phenomena which maybe be transient but important. For example, especially in a small spherical organism such as yeast, it may appear to be the case that a structure or aggregate has juxtanuclear localization, whereas 4D imaging may reveal that this is simply an artifact of the angle of inspection.
In the example experiment we present here, we demonstrate the use of a model misfolded protein, Ubc9ts, to follow aggregation quality control over time and space in the cytosol. At the permissive temperature, Ubc9ts is folded and diffused in the nucleus and cytosol. Upon heat-induced misfolding, it initially forms rapidly diffusing small aggregate Stress Foci that are processed for proteasomal degradation. When the proteasome is partially inhibited, these Stress Foci are converted into JUNQ and IPOD inclusions over the course of about 2 hr. If ubiquitin-mediated degradation is not available as a quality control option, Ubc9ts is immediately re-routed to the IPOD inclusion for protective aggregation. These tools offer incredible opportunities to discover novel genetic factors involved in aggregation quality control, and to explore their spatial and temporal regulation in the cell.
Offenlegungen
The authors have nothing to disclose.
Materials
| Name of the reagent | Company | Catalogue number | |
| MG132 | Mercury | mbs474790 | |
| con A | Sigma | C2010 | |
| Glass bottom plates | ibidi | ibd81158 | |
| 4D Fluorescence Imaging of Protein Aggregation Confocal 3D movies were acquired using a Nikon A1R-si microscope equipped with a PInano Piezo stage (MCL), using a 60X water objective NA 1.27, 0.3 micron slices, 0.5% laser power (from 65 mW 488 laser and 50 mW 561 laser). z-stacks were acquired every 5 min for 90 min. Each z-series was acquired with 0.5 micron step size and 30 total steps. Image processing was performed using NIS-Elements software. |
Referenzen
- Gershenson, A., Gierasch, L. M. Protein folding in the cell: challenges and progress. Current opinion in structural biology. 21, 32-41 (2011).
- Aguzzi, A., Calella, A. M. Prions: protein aggregation and infectious diseases. Physiological reviews. 89, 1105-1152 (2009).
- Morimoto, R. I. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes & development. 22, 1427-14 (2008).
- Cohen, E., Dillin, A. The insulin paradox: aging, proteotoxicity and neurodegeneration. Nature reviews. Neuroscience. , (2008).
- Hershko, A., Ciechanover, A. The ubiquitin system. Annual review of biochemistry. 67, (1998).
- Tyedmers, J., Mogk, A., Bukau, B. Cellular strategies for controlling protein aggregation. Nature reviews. Molecular cell biology. 11, 777-788 (2010).
- Treusch, S., Cyr, D. M., Lindquist, S. Amyloid deposits: Protection against toxic protein species?. Cell cycle (Georgetown, Tex.). 8, 1668-1674 (2009).
- Spokoini, R., et al. Confinement to Organelle-Associated Inclusion Structures Mediates Asymmetric Inheritance of Aggregated Protein in Budding Yeast. Cell Rep. , (2012).
- Weisberg, S. J., et al. Compartmentalization of superoxide dismutase 1 (SOD1G93A) aggregates determines their toxicity. Proc Natl Acad Sci U S A. 109, 15811-15816 (2012).
- Kaganovich, D., Kopito, R., Frydman, J. Misfolded proteins partition between two distinct quality control compartments. Nature. 454, 1088 (2008).
- Betting, J., Seufert, W. A yeast Ubc9 mutant protein with temperature-sensitive in vivo function is subject to conditional proteolysis by a ubiquitin- and proteasome-dependent pathway. The Journal of biological chemistry. 271, 25790 (1996).
- Tongaonkar, P., Beck, K., Shinde, U. P., Madura, K. Characterization of a temperature-sensitive mutant of a ubiquitin-conjugating enzyme and its use as a heat-inducible degradation signal. Analytical biochemistry. 272, 263 (1999).
