1. Programming by Early Life Adversity
Maternal separation (MS) is performed to induce early-life stress (ELS) in pups delivered by timed-pregnant C57BL/6N mice (postnatal day 0 (P0) on day of birth).
- Individual litters are placed in clean cages (with heating pad) for 3 h daily from P1-10.
- Control (non-ELS) pups remain undisturbed in the maternal nest throughout.
- Pups are kept with their mothers until weaning (P21), after which time they are housed in sex-matched groups (3-5 mice per cage) under standard laboratory animal housing conditions.
2. Isolation and Dissection of Brain Tissue
- Mice are killed at desired ages by cervical dislocation. Note: because this protocol involves a stress paradigm, no anesthesia is given prior to cervical dislocation, to avoid interfering with normal physiological regulation of stress hormones. Skulls are opened and brains are carefully removed and immediately snap-frozen by immersion in iso-pentane-dry ice, before being stored at -80 °C.
- Brains are cryosectioned (10 μm thickness); sections are mounted on Superfrost glass slides and maintained at -20 °C. Reference to a standard mouse stereotaxic atlas (e.g. Paxinos 6) is used to verify anatomical precision and to ensure inclusion of the region of interest (e.g. for neurons in the paraventricular nucleus – PVN – collect sections starting at the level of the rostral PVN, bregma -0.75 to -0.85).
- Sections are stained with cresyl violet to facilitate identification of different brain structures. Punches (0.8 mm) of the regions of interest (e.g. PVN) are obtained by in loco microdissection.
3. Nucleic Acid Extraction from Brain Punches
An optimized protocol for simultaneously extracting DNA and RNA from tiny neuroanatomically-defined brain regions for bisulfite and gene expression analyses is described 7.
Note: Considering that RNA is less stable than DNA in the GTC-Buffer, we recommend processing RNA first. The homogenate used for DNA purification can be kept at room temperature (RT) during that time.
- Homogenize punches using a pipette and vortexer in 400 μl of guanidinium thiocyanate (GTC) buffer (4.5 M guanidinium thiocyanate, 2% N-lauroylsarcosine, 50 mM EDTA pH 8.25 mM Tris-HCl pH 7.5, 0.1 M beta-mercaptoethanol, 0.2% antifoam A) at RT, passing several times through a hypodermic syringe (29G) (Figure 2).
- Split lysate into equal parts; both RNA and DNA may be extracted at the same time or separately, depending on particular experimental needs.
- For RNA purification add 1/10 volume of NaOAc, 1 volume AquaPhenol (Appligene) (pH 4) and 1/2 volume of chloroform:isoamyl (24:1) to lysate. Vortex vigorously after each step and incubate on ice for 10 min, centrifuge (20 min at 10,000 g at 4 °C); add an equal volume of 70% EtOH to aequous phase.
- Transfer mixture to an RNA spin column (e.g. Nucleospin RNA II from Macherey-Nagel) and perform on-column DNase-digestion and washing steps (follow manufacturer’s protocol); elute RNA in 25 μl H2O.
- For DNA purification an optimized protocol of the Qiagen DNeasy Blood and Tissue Kit is used. Equilibrate lysate with equal volumes of Buffer AL and 100% EtOH, load on a Spin Column centrifuge (1 min at 10,000 g at RT) and discard flow-through.
- Add 500 μl Buffer AW1 including 5 μl RNAse (1 mg/ml) to column and incubate 10 min at RT. Spin (1 min at 10,000 g, RT) and discard flow-through.
- Wash column with 500 μl Buffer AW2, discard flow-through and spin-dry empty column (1 min at 15,000 g).
- Add prewarmed (70 °C) Buffer AE and incubate columns for 10 min at 70 °C. Elute by centrifugation (1 min, 10,000 g), re-apply flow-through and repeat centrifugation step.
Use a spectrophotometer to determine DNA and RNA concentrations. A typical PVN punch yields ~600 ng DNA and ~400 ng RNA. For gene expression analysis by quantitative PCR (qPCR), we typically use ~100 ng of RNA in the reverse transcription reaction.
4. Bisulfite Conversion
Sodium Bisulfite is used to convert non-methylated cytosines to uracils. In contrast, methylated cytosines are protected from conversion. Therefore, all cytosines that are detected in the final sequencing of the bisulfite PCR-amplicon represent methylated cytosines.
Bisulfite conversion causes substantial DNA degradation which can be a limiting factor in PCR analysis. Optimized reaction conditions that maximize cytosine conversion, reduce DNA fragmentation and maintain single-stranded DNA even at lower temperatures can be achieved using Qiagen’s EpiTect Bisulfite Kit. In our hands ~200 ng of DNA purified from micropunches provide a sufficient amount of starting material for the bisulfite reaction.
To prevent differences in the efficiency of the conversion reaction, the amount of template DNA should be kept constant among all samples processed during an experiment. In addition, sequence reads should be scrutinized for conversion efficiency by examining the rate of non-converted non-CpGs. This rate should exceed 98%. Lower values indicate incomplete or inefficient bisulfite conversion and the underlying sequences should be excluded from the analysis.
5. Bisulfite PCR
- Design bisulfite sequencing primers that are specific to bisulfite converted DNA (see discussion); Methyl Primer Express software (https://products.appliedbiosystems.com/ab/en/US/adirect/ab?cmd=catNavigate2&catID=602121) will aid this and help to determine optimal annealing temperatures in pilot experiments.
- Prepare PCR-Master-Mix (for one reaction) as follows:
2.5 μl 10x PCR buffer
0.5 μl 10 mM dNTPs
1 μl 10 μM forward primer
1 μl 10 μM reverse primer
0.125 μl Qiagen Hotstart Taq Plus
fill up to 23 μl with H2O - Add 2 μl bisulfite-treated DNA to reaction.
- Amplify using the following conditions:
1 cycle 6 min 95 °C
45-50 cycles of 1 min 95 °C, 1 min at optimal annealing temperature, 1 min 72 °C
1 cycle 5 min 72 °C
Analyze 7 μl of PCR product by agarose gel electrophoresis to verify size of the amplicon and purify remaining PCR reaction for subsequent ligation using a commercially available PCR clean-up kit (e.g. Macherey-Nagel Nucleospin Extract). In case additional undesired PCR products are obtained, gel purification is recommended.
6. Bisulfite Sequencing
High-resolution DNA methylation profiles deduced from single clone readings can detect small changes in DNA methylation and identify regulatory regions that are responsive to treatment (environmental programming). The bisulfite sequencing process comprises three consecutive working steps. Firstly, PCR products obtained by prior bisulfite PCR are ligated into a vector and transformed into bacteria. Secondly, colony PCR from single clones is employed to determine the correct size of the insert. Thirdly, positive colony PCRs are cleaned up and subjected to Big-Dye sequencing reaction. Following a clean-up step, products are electrophoresed on a capillary sequencer.
6.1 Ligation and Transformation
Note: We routinely use the pGEM-T vector cloning kit (Promega). In our experience, the cloning efficiency depends critically on the insert to be ligated. Different vectors should be tested in case low numbers of recombinant clones are repeatedly obtained.
- Set up ligation reaction:
5 μl 2x Ligation Buffer
1 μl pGEM-T Vector
1 μl T4-Ligase
3 μl cleaned-up PCR product - Mix reaction by pipetting and incubate over night at 4 °C.
Note: Ligation may also be carried out for 1 h at RT as well. To increase number of recombinant clones, over night ligation at 4 °C is preferred. - Clean-up of ligation products by ethanol precipitation:
- Add 1 μl glycogen, 1 μl 3M NaAc and 25 μl 100% EtOH to the ligation reaction and precipitate for 1 min in liquid nitrogen.
- Centrifuge (15,0000 g for 30 min at 4 °C) and discard supernatant.
- Wash with 300 μl 70% EtOH and centrifuge (15,000 g for 20 min at 4 °C), discard supernatant and dry pellet at RT (10 min).
- Resuspend pellet in 10 μl H2O.
6.2 Transformation of ligation products in electrocompetent bacteria
- Precool electroporation cuvettes (1 mm width) on ice and thaw aliquot of electrocompetent DH5α bacteria on ice.
- Add 45 μl DH5α bacteria to 10 μl cleaned up ligation product (see above) and transfer to cuvette.
- Transform bacteria at 1.5 kV, 200 Ω, 15 μF and add 1 ml of prewarmed SOB medium directly after pulse delivery.
- Recover bacteria for 1 h at 37 °C, spread 100 μl of suspension on LB/ampicillin plates coated with IPTG/X-Gal.
- Incubate plates overnight at 37 °C.
6.3 Colony PCR
Note: Colony PCR from single clones is conducted to ensure that inserts are of predicted size. Delayed color development from the blue/white screening, undesired recombination events during ligation, incorporation of oligomeric primer pairs or truncated PCR products may otherwise lead to faulty sequencing results. We routinely use T7 and SP6 primers to amplify cloned inserts; this approach results in additional vector sequences of approximately 150 bp. T7 primer is used in the sequencing reaction in a later step.
- Set up colony PCR reaction in 96-well plate. For one reaction use:
3 μl 2.5 mM MgCl2
2.5 μl 10x Taq buffer
1.5 μl 10 mM dNTP
2 μl 2.5 mM T7 primer
2 μl 2.5 mM SP6 primer
1 μl Fermentas Taq Polymerase
fill up to 25 μl with H2O - Dispense 25 μl/well of master mix into each well of a 96-well plate.
- Pick positive (white) clone from plate with pipette tip and dip into PCR reaction.
- Amplify using following conditions:
1 cycle 4 min at 95 °C
10 cycles 30 s at 94 °C, 30 s at 56 °C and 30 s at 72 °C
30 cycles 30 s at 94 °C, 30 s at 48 °C and 30 s at 72 °C
1 cycle 5 min at 72 °C - Load 5 μl of colony PCR on an agarose gel and determine reactions that contain the right insert size.
- Colony PCRs containing the desired amplicons are cleaned up using a commercially available kit (Machery Nagel Nucleofast).
6.4 Big-Dye terminator reaction and sequencing
- Prepare Master mix for Big-Dye reaction. For one reaction use:
1 μl H2O
0.5 μl Big-Dye reagent
1.5 μl Sequencing buffer - Dispense 3 μl/well into each well of a 96-well plate, to each well add 2 μl of cleaned up colony PCR product and run reaction on a thermocycler with the following parameters:
1 cycle 1 min at 96 °C
35 cycles of 10 s at 96 °C, 5 s at 50 °C and 4 min at 60 °C - The Big-Dye reaction is cleaned up using a commercial kit (Millipore Montage 96 Sequencing Clean-Up Kit) and processed on a capillary sequencer (e.g. ABI 3100 DNA).
- Sequences are analyzed using the Biq Analyzer (http://biq-analyzer.bioinf.mpi-sb.mpg.de/) or the online tool BISMA (http://biochem.jacobs-university.de/BDPC/BISMA/) to derive the methylation pattern of the investigated DNA region (Figure 4).
7. Representative Results
To gain insight into the influence of ELS on the Avp expression and methylation status, C57BL/6N mice were processed according to the workflow described above. Briefly, a group of C57BL/6N mice was subjected to ELS while the control group was left undisturbed. The PVN and the supraoptic nucleus (SON) were punched and DNA and RNA were isolated simultaneously from the single punches. RNA was reverse transcribed and the levels of Avp transcripts were measured by qPCR analysis and normalized to the expression of Hprt and Gapdh housekeeping genes. DNA was bisulfite treated, amplified with primers specific to the Avp enhancer (Figure 4a) and the PCR products were cloned and sequenced. At least 20 recombinant clones from each mouse/PCR were analyzed to determine methylation frequencies for the CpGs contained in the PCR amplicon (Figure 4b). Compared to controls, ELS induced a significant hypomethylation at CpG10, CpG12, CpG13 and Cpg14 of the Avp enhancer (p < 0.05, n = 6-8 animals) suggesting epigenetic marking of this regulatory region through early-life experiences. In contrast to the PVN, methylation of the Avp enhancer was unaffected by ELS in the SON illustrating tissue specificity of epigenetic marking (Figure 4c). Analysis of the DNA methylation status at CpG10 and Avp gene expression in control animals (n = 6) evidenced a negative correlation pointing to a role of DNA methylation in the fine-tuning of AVP gene expression (Figure 4d).

Figure 1. Micropunches are obtained from dissected brains from control and early-life stressed mice. Following DNA and RNA isolation, gene expression is determined by qRT-PCR while bisulfite treated DNA is amplified and purified products are cloned in a suitable vector allowing identification of recombinant clones by blue/white selection. Correct insert sizes are verified by colony PCR prior to carrying out Big-Dye reaction and processing on a capillary sequencer. Methylation pattern are visualized by appropriate software tools. Click here to view larger figure.
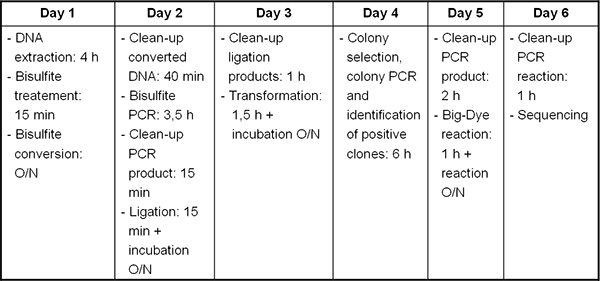
Figure 2. Timeline from DNA/RNA extraction to bisulfite sequencing.
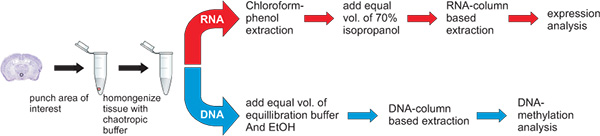
Figure 3. Workflow for the simultaneous extraction of DNA and RNA. The punch of the hypothalamic nucleus paraventricularis, a tiny Avp gene expressing brain region, is placed in 400 μl GTA buffer, vortexed until disrupted and further homogenized by passing through a syringe (29G). The homogenate is then split for the purification of RNA and DNA. The split can be 1:1 or of different proportions depending on the particular experimental question. Homogenates should be processed within a few hours.
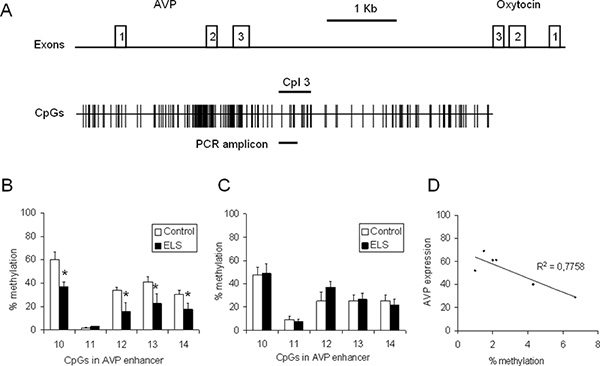
Figure 4. CpG methylation at the Avp locus in 6 weeks old control and ELS animals. (A) Scheme of the Avp and oxytocin genes orientated tail to-tail and separated by the intergenic region (IGR). Exons are depicted by open (numbered) boxes. The distribution of CpG residues is indicated and size and position of the respective PCR amplicon containing CpG 10 to CpG14 is marked by a bold line. (B) Methylation of the Avp enhancer in the PVN in control and ELS animals (n = 6-8). (C) Methylation of the Avp enhancer in the SON in control and ELS animals (n = 8-10). (D) Avp gene expression was correlated with methylation at CpG10 (n = 6). Click here to view larger figure.
