The purpose of this video article is to provide an overview of the work flow along with data format and interpretation for researchers that may just be starting this type of work (Figure 1). It includes demonstration of techniques, simplified protocols and practical tips described in previously published works.5,10
General Work Flow and Laboratory Facilities:
There should be at least 3 separate work areas for this kind of research. The laboratory should have 1) a pre-PCR area with a PCR hood (clean air box or isolated work area) for primer preparation (dilution, aliquot preparation and mixing), 2) a separate bio-safe cabinet for handling and addition of DNA to the PCR mixtures, and 3) a post-PCR work area for preparing and loading gels and for preparing samples for FLA. Primers and DNA samples should be kept in separate freezers and refrigerators. Primer contamination is one of the chief and most persistent problems in laboratory work of this type. Pipettes used for primers and PCR mixes should NOT be used for DNA. There should be separate sets of pipettes for the pre-PCR, PCR and post-PCR work areas. Generally, one researcher can process 12-18 samples in a 12-24 hour period using standard laboratory equipment.
Before beginning each phase of work:
- Wear a lab coat and gloves. Gloves should be worn any time biological samples, primers, DNA and ethidium bromide are being handled. Change gloves frequently.
- Work in a PCR cabinet hood, biosafety cabinet or clean work area.
- Use a fresh bench paper or pad.
- Set up waste receptacles suitable for liquids and pipette tips.
- Wipe down with the inside of the PCR hood and pipettes with 70% ethanol.
- Subject pipettes and work area to UV light for 15 minutes before PCR set up. (Ultraviolet light crosslinks surface DNA contaminants.)
- Always use aerosol prevention pipette tips for materials containing DNA, primers and PCR reagents.
- Centrifuge any tubes/strips/plates containing liquid before opening to prevent aerosol escape and cross-contamination by handling.
1. M. leprae DNA preparation
Clinical samples containing M. leprae are obtained from leprosy patients who visit skin clinics. Routine diagnostic samples may be skin punch biopsies, slit skin smears or nasal swabs. Generally, punch biopsies or slits skin smears are the best for molecular epidemiology because they are clean and contain sufficient amounts of M. leprae. Use of these materials for research must be approved according to institutional guidelines.
- Preserve biopsy or slit skin smear samples in a screw-cap vial with 1 ml of 70% ethanol.
- Centrifuge each sample in a variable speed bench-top centrifuge at 12,000 x g for 15 minutes.
- Remove the supernatant and place it in a microcentrifuge tube. If necessary, it may be useful to re-centrifuge these samples and recover additional tissue or DNA later.
- Add 500 μl of phosphate buffered saline (PBS) to the tissue samples and soak for 1 hour to exchange residual ethanol preservative and rehydrate the sample.
- Centrifuge the samples in a bench-top centrifuge at 12,000 x g for 20 minutes. Discard the PBS solution in a waste container partially filled with disinfectant solution.
- Extract bacterial DNA using the Qiagen DNEasy Blood and Tissue kit following the guidelines prescribed.
- Extracted DNA should be aliquoted into 4 vials. Store 2 aliquots at -80°C for future use if/when reproducibility of test results is required or when new technologies become available. Store one aliquot at -20°C and one at 4°C for more immediate use.
- It is prudent to prepare an ‘extraction blank’ along with the tissue samples. The blank is subjected to all the same treatments outlined above but is performed without tissue. PCR the extraction blank along with the patient samples to guarantee that the operator’s extraction technique is correct and that reagents are free of DNA contamination.
2. Primer preparation
- Primers for the loci for which there are the most extensive strain type data are listed in Table 1. Four or 5 primers are combined for multiplex PCR. One primer for each locus carries a 5′ fluorescent chemical tag that will be detected during capillary electrophoresis fragment length analysis (FLA).
- For each multiplex PCR combination, the tagged primer and corresponding reverse primer for each locus are combined in separate Eppendorf tubes: forward primers in one tube, reverse in another. Stock primers are prepared as 100μM solutions (Figure 2a), a portion of which is diluted 10x to a concentration of 10 μM using TE (1x Tris-EDTA, pH 8.0). Remaining aliquots of the 100 μM primer solutions are stored at -20°C for later use.
- Equal quantities of each 10 μM primer are mixed resulting in 2μM final concentrations of each primer. When the primer combinations are added to the PCR mixture (Table 3), the final concentration of each primer drops to 0.2 μM.
When the combined primers are added to the PCR mixtures (Table 3), final concentrations drop to 0.2μM each.
3. Amplifying bacterial DNA using multiplex PCR
- PCR Setup
- Record the number of samples that will be used and prepare a worksheet with the required reagents and their volumes before collecting the necessary plastics and other materials.
- Label the sterile tubes/strips/plates to be used for the PCR with sample numbers. Remember to include positive and negative controls.
- Wipe down the thermocycler with a cleaning solution (such as Decon ELIMINase) and program it according to Table 2. Change gloves after cleaning and before handling primers and other reagents.
- Prepare PCR Mastermix according to Table 3 and label the PCR tubes/strips/plate with the primer combination and sample numbers to be used.
- Aliquot 18μl of Mastermix to each labeled PCR sample tube or well. Change gloves.
- Wipe down a separate bio-safety cabinet and pipettes with 70% ethanol to help clean and disinfect the work area and tools, then subject the work area and pipettes to UV light for 15 minutes to cross-link any DNA contaminants. Change gloves.
- In the clean bio-safe cabinet, add 2μl of the DNA template to the Mastermix in each PCR tube/plate well to acquire a total volume of 20μl (Figure 2b). Use aerosol prevention pipette tips for all liquid materials.
- Centrifuge the PCR tubes/strips/plates briefly to mix contents.
- Place the sample tubes/strips/plate in the thermocycler and start the PCR program.
- When the program is complete, remove the products from the thermocycler and store at 4°C until electrophoresis.
4. Gel electrophoresis of PCR products
*This protocol has been standard for many years and is an optional step that can be employed if confirmation of PCR products is desired prior to sending them for FLA.
- In a separate post-PCR work area, prepare a 2% agarose gel. Use 2.0g agarose powder/100ml of 1x TBE (Tris/Borate/EDTA) buffer solution in a flask. Heat the mixture for about 1.5-2 minutes in a microwave oven. Swirl the contents, heating again if necessary to dissolve the agarose. Cool slightly and pour the gel in a form using a comb with enough wells for the samples.
- Remove the PCR products from 4°C and centrifuge for about 30 seconds.
- Mix 2-5μl of PCR product with 0.5-1μl of 5x or 6x gel loading buffer (Loading buffer is available from various companies, or may be mixed up in the lab. Recipes are available online.)
- Load the wells of the gel with the 6μl sample/loading buffer mix. Add a molecular ladder to one well (preferably a 20 bp ladder).
- Run the gel at 100V for approximately 90 minutes.
- Soak the gel in ethidium bromide solution for 15-30 minutes, then in ultrapure distilled water for an equal amount of time.
- Image the gel while UV light is applied. There should be a band for each of the 4-5 DNA segments in the combination (Figure 3).
- Dispose of the gel in accordance with the institution’s hazardous materials policy. Ethidium bromide solution can be used multiple times before proper disposal.
5. Preparing samples for fragment length analysis (FLA)
- In a clean Eppendorf tube, prepare a master mixture containing 12μl of Hi-Di formamide solution from a fresh aliquot and 0.3μl of GeneScan -500 LIZ sizing standard (both from Applied BioSystems) for each sample to be tested. (Hi-Di formamide chemically denatures the DNA strands prior to capillary electrophoresis, eliminating the need for heating.) Using a 96-well optical quality reaction plate, aliquot 12.3μl of the formamide-LIZ mix to each well being used for samples and set the plate aside.
- Make a plate map for your records indicating which DNA sample is in each well (Figure 4a).
- Using tubes/strips or a 96-well plate, add 1μl of PCR product (from Part 3) to 59μl of PCR quality water creating a 1:60 dilution of the PCR product. Dilutions may be adjusted based on signal strength from the FLA data or brightness of gel bands. Even lower concentrations of DNA may be sufficient (1:120 or 1:180).
- Add 1μl of the diluted PCR product to the correct well in the plate containing the formamide-LIZ mixture.
- Speed and efficiency can be greatly enhanced if the PCR was also done in a 96-well plate. One can simply line up 3 such plates in sequence: Plate 1 with PCR products, Plate 2 with sterile water for the dilution of the PCR products, and Plate 3 with formamide and sizing ladder for fragment length analysis. A multichannel pipette allows for a quick process of FLA preparation.
Sample Analysis via Genetic Analyzer
- Calibrate the Genetic Analyzer to detect the applied flurophores per the manufacturer’s instruction. Be sure to use a dye-set that includes LIZ.
- Replenish the water rinse containers and add new Running Buffer with EDTA (1x) (Applied BioSystems) to buffer chambers.
- Add a pre-slit silicon septum to the plate and place the plate in the designed holding tray. Press the ‘Tray’ button on the Genetic Analyzer to bring the autosampler forward. Place tray onto autosampler and close the door to the Genetic Analyzer.
- Create or import a spreadsheet for the analysis. Importable files have a .plt extension and are in tab-delimited format. Files can be modified in Excel (Microsoft) or similar programs.
- The samples are injected into the capillary (50-cm length, POP-7 polymer) by applying an injection voltage of 1.6 kV for 15 s. The capillary electrophoresis runs at a voltage of 15 kV at 60°C for 1800 seconds. The entire process takes about 45 minutes.
- Once a run is complete, the data for each sample analyzed are converted into files with a .fsa extension and placed in a plate file folder (Figure 4b). Each data file is roughly 100 kB in size and can be stored on a flash drive or zipped and emailed. Data files can be viewed using suitable software, such as ABI’s GeneMapper or Peak Scanner.
6. Analysis of fragment length results
- Analysis of the data from fluorescent capillary electrophoresis requires special software. If such software is not available, go the Applied BioSystems website and download Peak Scanner. The software is free and works quite well. https://products.appliedbiosystems.com/ab/en/US/adirect/ab?cmd=catNavigate2&catID=603624
- Open Peak Scanner and ‘Start New Project’ followed by ‘Add Files’. Load the selected .fsa data files into the program, including the positive and negative controls.
- Select (highlight) and “analyze” all loaded files. Be sure each sample is set to “Size Standard: GS500(-250) and Analysis Method: Sizing Default-pp. Press ‘Analyze’.
- Examine the size in base pairs of each colored peak produced by the DNA fragments in the samples.
- Record each peak size value in base pairs and compare it to the positive control peak.
- Peaks in the positive control samples should compare favorably with the numbers of base pairs and corresponding numbers of tandem repeats listed in Tables 4-7.
- Determine how many short tandem repeat segments are present in each sample allele by comparison with the positive control. We use NHDP63 which has been sequenced for the number of VNTR copies at each locus being examined.4
- Enter the recorded data into a spreadsheet for comparative and/or mathematical analyses. VNTR ‘fingerprints’ or haplotypes, are strings of alleles at defined loci that are characteristic of a M. leprae strain.
7. Representative Results
Gel electrophoresis of the PCR products will hopefully produce a band for each locus in the primer combination (Figure 3). In Figure 3, there are 2 sections to the gel: the top section has Combination 1 PCR samples and the lower portion Combination 2 samples. Each section contains a 20 base pair molecular ladder, followed by PCR products obtained from 8 patient samples. Combination 1 also has a negative control and finally a positive control (NHDP63 strain). (The controls for combination 2 were on a different gel.) Note that most samples clearly display 5 bands, 1 for each locus in the combination. In some cases, bands may be too close together to appear as separate loci resulting in the appearance of only 4 bands.
Expected amplicon sizes are listed in Table 1. Our laboratory uses NHDP63 strain of M. leprae as a positive control. Two types of repeat segments have been studied: microsatellite loci (with 1-5 base repeats) and minisatellite loci (with segments greater than 5 base pairs repeated multiple times). 5
Interpretation of the data files from capillary electrophoresis relies on 2 standards: an internal DNA fragment sizing standard called GeneScan -500LIZ (ABI) and an external positive control sample of amplified bacterial DNA.
The FLA chromatograms, viewed using Peak Scanner software, can be seen in Figures 5a, 5b and 6.
Peak Scanner provides data on amplicon size (x-axis in base pairs) and signal strength (y-axis). (Figure 5b) Additional data on peak area, etc. are also available, although the size and peak height values are most important. Peaks less than 100 units in height are usually considered too weak a signal to be reliable.
Figure 6 compares the positive control (NHDP63) and two patient samples, showing a variation in the number of tandem repeats at locus (GTA)9. In the positive control, the sequence (GTA) is repeated 10 times. The NHDP63 VNTR and amplicon size were verified through gene sequencing. Patient 4’s PCR amplicon is 3 bp smaller than the positive control indicating there are only 9 repeat units, while Patient 6 has an amplicon that is 3 bp larger than NHDP63 revealing 11 repeats of (GTA). Patient 2 had low bacterial index (BI) with little or no DNA PCR replication, therefore no FLA signal.
Difficulty in FLA data interpretation sometimes occurs as a result of ‘stuttering’. During the PCR reaction, the DNA polymerase may produce fragments that are 1 or more repeats longer or shorter than the source allele. These are usually recognized as peaks of lower height surrounding the main peak. They will be ‘on the ladder’ that is, the correct number of base pairs of the repeat segment larger or smaller than the principal peak. The result is a family of peaks of the same color. Figure 7 shows this with locus (TA)10.
Another difficulty sometimes encountered with FLA involves ‘+A’ or ‘A-tailing’, in which the DNA polymerase adds a single base [usually adenine (A)] to the 3′ end of the copied DNA segment. This shows up in FLA as a peak to the right of a main or stutter peak that is 1 base pair larger than the adjacent peak as shown in Figure 8. It should not be confused with a main or stutter peak. A principal peak and its A-tail are considered a single species. The A-tail does not alter the number of VNTR repeats. (Some DNA polymerase kits are designed to specifically promote A-tailing in order to reduce this confounding effect. Promoting complete A-tailing tends to produce a single peak rather than a pair of peaks.)
The DNA extraction and PCR products contain both M. leprae and human DNA, however with the exception of (TA)18, the primers are specific enough that they only amplify the bacterial DNA, and there is little or no human DNA amplification. (TA)18 often produces a peak at 242 base pairs that is from human DNA and is not seen in armadillo passaged DNA samples (Figure 9).
The shelf life of primers is generally good provided they are kept at -20°C, only removed for immediate use, and then stored at 4°C until consumed. Primers should be stable at 4°C for 1-2 weeks. Even though making primer combinations for PCR is somewhat time consuming, it is recommended that only enough primer combination for immediate use be prepared. Primer combinations seem to degrade somewhat when stored for prolonged periods.
TE should be aliquoted from larger stocks, and aliquots may be stored either frozen or at room temperature. Larger aliquots of 200-400μl are good for diluting dried or concentrated primer stocks (Figure 2a). Smaller aliquots of 10-50 μl are useful for supplementing the volumes of primer combinations 3 and 4. TE is inexpensive and aliquots should be discarded after use.
Multiplex enzyme kits are quite expensive and should be kept frozen (-20°C) until use. Following PCR preparation, any unused multiplex solutions should be returned immediately to 4°C. The small Qiagen Multiplex PCR Kit comes with 3 tubes of multiplex mix, each containing 0.85ml (850μl) of solution; enough for about 65-70 PCRs.
Generally, it is best to avoid repeated, major temperature changes for the materials used in this type of laboratory work including the DNA samples, primers and multiplex kit solutions. All materials should be stored at -20°C until needed, and then kept at 4°C until consumed. Long term storage of DNA should be at -80°C.
All materials containing spent tissue, primers and/or DNA should be autoclaved and disposed of when no longer of any use. All samples treated with ethidium bromide or formamide should be treated as hazardous waste and disposed in accordance with the institution’s hazardous materials policy.
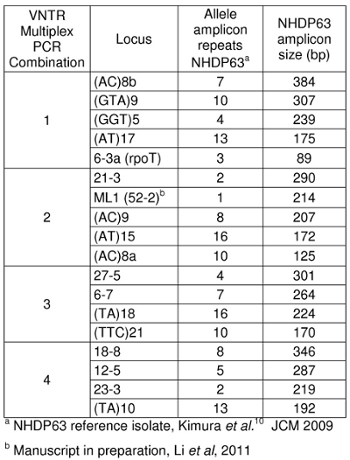
Table 1: Amplicon sizes for strain NHDP63
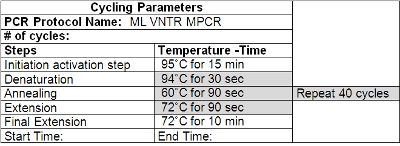
Table 2: Cycling Parameters for VNTR PCR

Table 3: Preparation of PCR
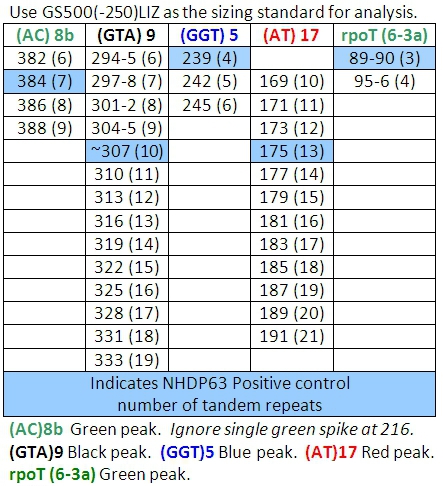
Table 4: Allele Calls for Combination 1
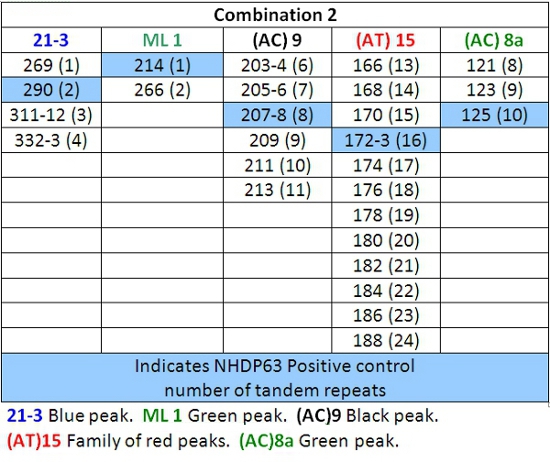
Table 5: Allele Calls for Combination 2
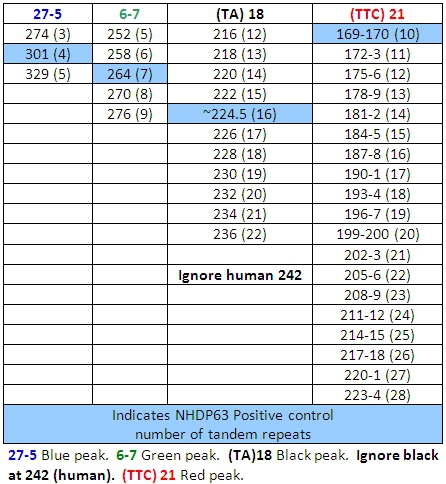
Table 6: Allele Calls for Combination 3
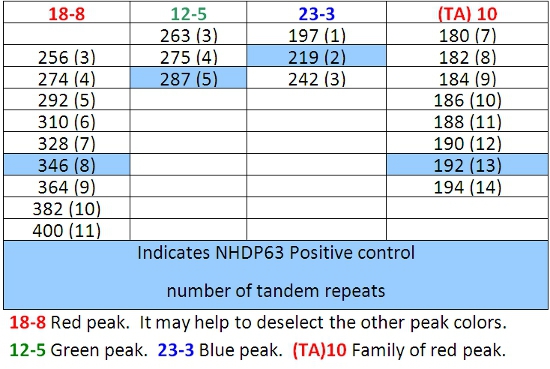
Table 7: Allele Calls for Combination 4
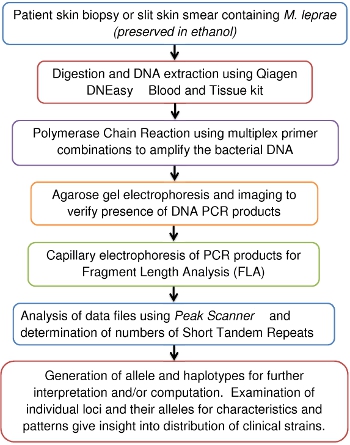
Figure 1: VNTR-FLA Process Flow Diagram

Figure 2. (a) Preparation of Combination 1 Upper Primers (Eppendorf tube picture courtesy of www.clker.com). (b) PCR Setup (for 8 PCRs) (Eppendorf tube picture courtesy of www.clker.com)
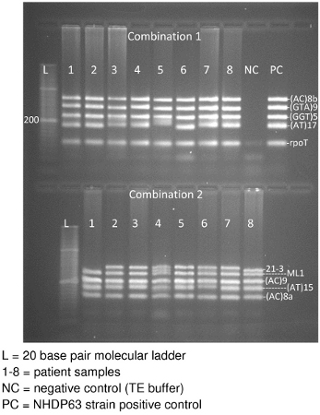
Figure 3. Agarose gel of Combinations 1and 2 VNTR PCR product DNA
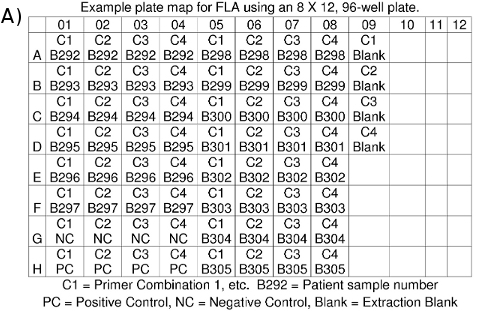
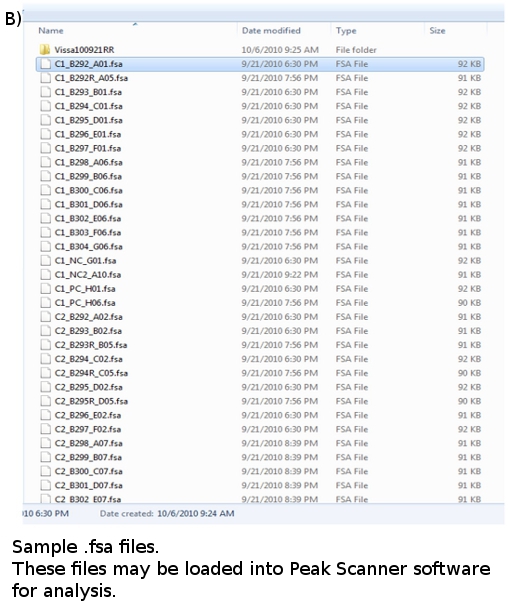
Figure 4. (a) FLA Plate Map. (b) FLA data files: *.fsa

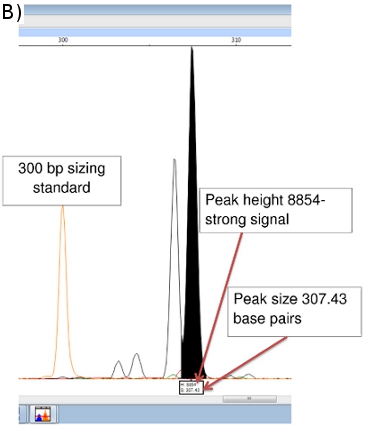
Figure 5. (a) FLA Chromatogram of Positive Control (NHDP63) for Combination 1 VNTR loci. (b) Peak Scanner data for amplicon size and abundance of (GTA)9
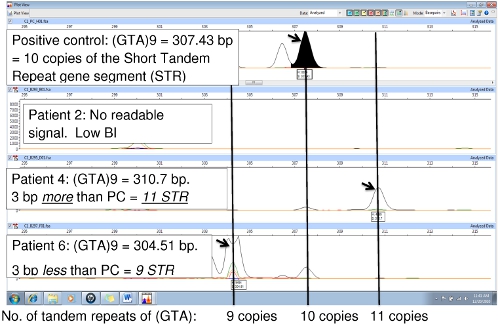
Figure 6.Comparison of PCR samples to the PC (NHDP63) for locus (GTA)9
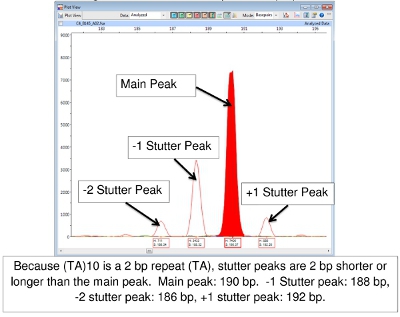
Figure 7.Main and Stutter Peaks for (TA)10

Figure 8: +A (A-tail) Peaks Adjacent to Main or Stutter Peaks.
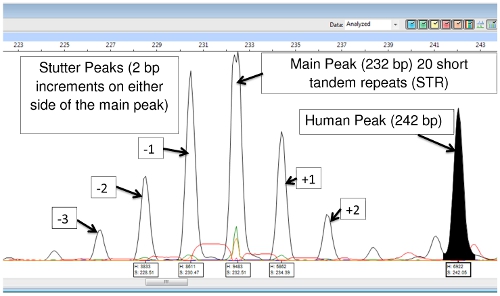
Figure 9: (TA)18 Main Peak, Stutter Peaks and Human DNA Peak

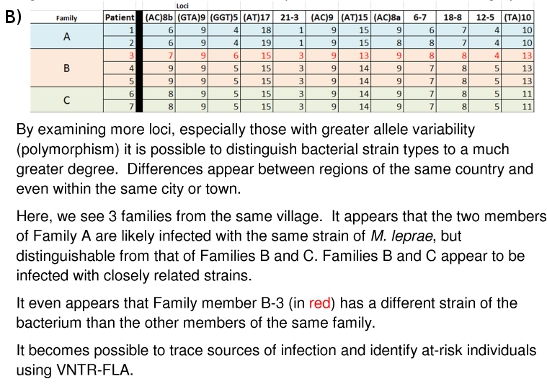
Figure 10: (A) Strain Differentiation of M. leprae based on VNTR data. (B) Strain Differentiation of M. leprae based on MLVA DNA Fingerprint
