1. Loading and Running the Gel
- All samples must be prepared adequately to maintain the function of the enzymes and used immediately after collection or stored frozen at -80°C. The samples must not contain reducing agents (such a β-mercaptoethanol) or be boiled before gel loading.
- Open the pouch containing a gel inside a cassette, rinse the cassette with deionized water.
- Remove the protective tape from the bottom of the cassette and the comb from the top of the gel.
- Rinse the wells three times with running buffer (diluted to 1X in deionized water).
- Place the gel into the Mini-Cell, ensuring that the smaller side of the cassette faces inwards. Lock into place with the Gel Tension Wedge. The Mini-Cell allows to run one or two gels in parallel.
- Fill the top (inside) chamber with 1X running buffer above wells level, check for any leaks. In case of leaks, remove the buffer and reposition the gel.
- Fill the lower chamber with 1X running buffer.
- Load 10 μL of protein molecular marker in one well.
- Mix an equal amount of gel-loading buffer and of sample and load into the wells of the gel using gel-loading tips (changed between each sample). These wells can be loaded with up to 20 μL total.
- Place the lid on the Mini-Cell and connect the electrode cords to the power supply. Switch the power supply on and set it to run at 125 V constant for 90 minutes. Check the formation of small bubbles on the wire of the lower chamber, indicating current circulation.
- When running your first gel, monitor progress of the migration every 15 minutes, using the bromophenol blue included in the loading buffer as an indicator. Let the gel run until the indicator dye reaches the bottom of the gel.
2. Renaturing and Developing the Gel
- For each gel, prepare 100 mL of 1X renaturing buffer and 200 mL of denaturing buffer, both in deionized water.
- When the bromophenol blue tracking dye reaches the bottom of the gel, switch the power supply off, open the Mini-Cell, and remove the gel. Separate the two sides of the cassette using the gel knife (or a weighing spatula). Cut a corner to mark the gel’s direction.
- Carefully remove the gel from the cassette and place into a container with 100 mL renaturing buffer. Incubate for 30 minutes at room temperature with gentle agitation.
- Remove the renaturing buffer and add 100 mL of developing buffer to the gel. Incubate for 30 minutes at room temperature with gentle agitation.
- Remove the developing buffer and add 100 mL more of developing buffer to the gel. Incubate overnight (16-18 hours) at 37°C.
- Remove the developing buffer and rinse three times (5 minutes each) with deionized water under gentle agitation at room temperature.
- Scan the gel to save the exact positioning of the protein standard bands as they will become less or not visible after gel staining.
- Stain the gel by adding 20 mL of SimplyBlue Safestain to the gel. Incubate for 1 hour at room temperature under gentle agitation.
- Remove the SimplyBlue SafeStain and de-stain the gel in 100 mL or more deionized water for one hour at room temperature under gentle agitation.
- For better results, replace with fresh deionized water and incubate for another hour or more at room temperature under gentle agitation.
3. Data Analysis
- Carefully remove the gel from the water and place in a plastic sheet protector.
- Scan the gel with a resolution of 300 dpi or higher. Save the image in the TIFF format (Figure 1A).
- Measure band intensities with ImageJ (or another similar software).
- Open the TIFF file in ImageJ (Figure 1A).
- Visualize in black and white by selecting “Image>Type>8-bit” (Figure 1B).
- Use the rectangular selection tool to outline the first band, drawing a rectangle at least twice higher than it is wide (this is a software requirement).
- Press “1” or select “Analyze>Gels>Select first lane” and the band will be outlined.
- A new rectangle will appear, move it to the next band and select “Analyze>Gels>Select next lane”. Repeat until all bands are selected (Figure 1B).
- Press “3” or select “Analyze>Gels>Plot lanes” to generate the profile plot for each band (Figure 1C).
- Use the straight line selection (should already be automatically selected) to draw base lines so that the peak of interest is a completely enclosed area.
- Select the wand tool and click inside each peak to select it.
- Select “Analyze>Gels>Label peaks” to obtain a table with the area for each selected peak. These data can be plotted as such (Figure 1D) or can be normalized to the value of a chosen band. This normalization is especially important when pooling values from several replicate gels to determine statistical significance of the results.
4. Representative Results
We show a gel with 11 wells loaded with different cell culture supernatants containing different amounts of MMP-2 (Figure 1A). Direct observation of this gel shows obvious differences in MMP-2 concentrations between some of the wells. For example, it is clear that wells # 4 and 5 contain much more MMP-2 than well # 11 or even well # 10. For objective quantification of bands we have used densitometry with the ImageJ software (Figure 1B-D), confirming an approximately 4-fold difference in MMP-2 amounts between well # 11 and wells # 4 and 5 and an approximately 2-fold difference in MMP-2 amounts between well # 10 and wells # 4 and 5.

Figure 1. Detection of MMP-2 by gelatin zymography. A, Scanned image of a gelatin gel. Well numbers are indicated at the top of the gel. B, Same gel as in A shown in black and white for densitometry. Each band was selected with a rectangle in ImageJ. C, Two examples of densitometry profiles for the gel shown in A and B (bands # 4 and # 11). A straight line was drawn at the base of each peak to create a closed area. D, Plot of the peak areas for the gel shown in A and B before (left) and after (right) normalization to the density of band # 1.
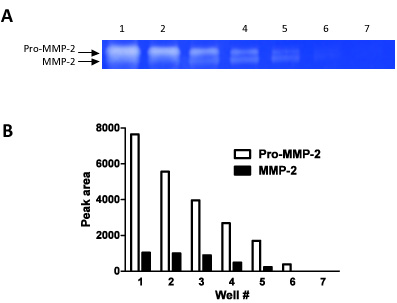
Figure 2. This figure demonstrates how zymography can be used as a semi-quantitative technique. We have loaded serial dilutions (steps of 1:1 dilutions) in 7 consecutive wells and measured the band densities for both pro-MMP-2 and MMP-2.
